REKLAMA
Akty ujednolicone - rok 2013 poz. 1
ROZPORZĄDZENIE WYKONAWCZE KOMISJI (UE) NR 503/2013
z dnia 3 kwietnia 2013 r.
w sprawie wniosków o zatwierdzenie genetycznie zmodyfikowanej żywności i paszy zgodnie z rozporządzeniem (WE) nr 1829/2003 Parlamentu Europejskiego i Rady oraz zmieniające rozporządzenia (WE) nr 641/2004 i (WE) nr 1981/2006
(Tekst mający znaczenie dla EOG)
KOMISJA EUROPEJSKA,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie (WE) nr 1829/2003 Parlamentu Europejskiego i Rady z dnia 22 września 2003 r. w sprawie genetycznie zmodyfikowanej żywności i paszy (1), w szczególności jego art. 5 ust. 7, art. 11 ust. 5, art. 17 ust. 7 i art. 23 ust. 5,
po konsultacji z Europejskim Urzędem ds. Bezpieczeństwa Żywności,
a także mając na uwadze, co następuje:
(1) W rozporządzeniu (WE) nr 1829/2003 ustanowiono unijne procedury zatwierdzania i nadzorowania genetycznie zmodyfikowanej żywności i paszy oraz znakowania takiej żywności i paszy. Rozporządzenie przewiduje przeprowadzenie naukowej oceny zagrożeń, które zmodyfikowana żywność lub pasza mogą stwarzać dla zdrowia ludzi i zwierząt oraz, w stosownych przypadkach, dla środowiska. Rozporządzenie stanowi także, że genetycznie zmodyfikowana żywność lub pasza nie mogą wprowadzać w błąd konsumenta albo użytkownika ani nie mogą różnić się od żywności lub paszy, którą mają zastępować, w stopniu, w którym normalna konsumpcja byłaby niekorzystna dla ludzi lub zwierząt pod względem odżywczym.
(2) W szczególności rozporządzenie (WE) nr 1829/2003 przewiduje, że wnioski o zatwierdzenie muszą w sposób odpowiedni i wystarczający wykazywać, że genetycznie zmodyfikowana żywność i pasza spełniają wymogi ustanowione w tym rozporządzeniu pod względem zamierzonego użycia.
(3) W interesie spójności prawodawstwa Unii niektóre definicje ustanowione w rozporządzeniu (WE) nr 178/2002 Parlamentu Europejskiego i Rady z dnia 28 stycznia 2002 r. ustanawiającym ogólne zasady i wymagania prawa żywnościowego, powołującym Europejski Urząd ds. Bezpieczeństwa Żywności oraz ustanawiającym procedury w zakresie bezpieczeństwa żywności (2) powinny być stosowane również do niniejszego rozporządzenia.
(4) Rozporządzenie Komisji (WE) nr 641/2004 (3) w sprawie szczegółowych zasad wykonywania rozporządzenia (WE) nr 1829/2003 przewiduje pewne szczegółowe zasady dotyczące wniosków o zatwierdzenie składanych zgodnie z rozporządzeniem (WE) nr 1829/2003. Aby ułatwić przygotowanie wniosków oraz zagwarantować, że zawierają wszystkie informacje niezbędne do ich oceny, należy przewidzieć bardziej wyczerpujące i systematyczne zasady dotyczące wniosków o zatwierdzenie, które powinny być także specyficzne dla typów organizmów zmodyfikowanych genetycznie, tj. roślin, zwierząt i mikroorganizmów.
(5) Przepisy ustanowione w niniejszym rozporządzeniu powinny obejmować jedynie wnioski dotyczące genetycznie zmodyfikowanych roślin przeznaczonych na żywność lub paszę, żywności lub paszy zawierających rośliny zmodyfikowane genetycznie lub składających się z nich, a także żywności lub paszy produkowanych z takich roślin. Znaczna większość obecnych wniosków dotyczy roślin zmodyfikowanych genetycznie, w przypadku których dostępne jest wystarczające doświadczenie.
(6) Przepisy ustanowione w niniejszym rozporządzeniu powinny precyzować ogólne wymagania dotyczące przedstawienia i przygotowania wniosków, tj. wymagania dotyczące dostarczenia informacji ogólnych i naukowych, obejmujących metody wykrywania, oraz identyfikacji, a także dostarczenia materiałów odniesienia, tak aby wnioski spełniały warunki ustanowione w art. 5, 17 i 30 rozporządzenia (WE) nr 1829/2003.
(7) Wnioskodawca powinien wziąć pod uwagę informacje naukowe, które należy dostarczyć we wniosku w odniesieniu do oceny ryzyka dla środowiska naturalnego stwarzanego przez organizmy zmodyfikowane genetycznie lub żywność i paszę zawierające organizmy zmodyfikowane genetycznie lub składające się z nich, zgodnie z zasadami dotyczącymi oceny ryzyka dla środowiska naturalnego w załączniku II do dyrektywy 2001/18/WE Parlamentu Europejskiego i Rady z dnia 12 marca 2001 r. w sprawie zamierzonego uwalniania do środowiska organizmów zmodyfikowanych genetycznie i uchylającej dyrektywę Rady 90/220/EWG (4). Wnioskodawca powinien również wziąć pod uwagę wytyczne publikowane przez Europejski Urząd ds. Bezpieczeństwa Żywności (EFSA) w tym zakresie.
(8) Oprócz wymagań ogólnych dotyczących przedstawienia i przygotowania wniosków należy przewidzieć szczegółowe przepisy gwarantujące, że informacje naukowe wymagane we wniosku w sposób odpowiedni i wystarczający wykazują, że genetycznie zmodyfikowana żywność lub pasza spełniają wymogi ustanowione w rozporządzeniu (WE) nr 1829/2003 w odniesieniu do zamierzonego użycia.
(9) Przepisy te powinny zatem wyszczególniać badania, które należy dołączyć do wszystkich wniosków, a także metody badawcze, które należy zastosować do przeprowadzenia tych badań, uwzględniając jednocześnie właściwe normy międzynarodowe, takie jak wytyczne Kodeksu Żywnościowego dotyczące przeprowadzania oceny bezpieczeństwa żywności pochodzącej z roślin o rekombinowanym DNA (5).
(10) Zgodnie z obowiązującymi wytycznymi EFSA (6) ocena bezpieczeństwa genetycznie zmodyfikowanej żywności lub paszy powinna zawierać badania dotyczące nowych składników wynikających z modyfikacji genetycznej, charakterystykę molekularną rośliny zmodyfikowanej genetycznie, analizę porównawczą składu i fenotypu rośliny zmodyfikowanej genetycznie w porównaniu z jej tradycyjnym odpowiednikiem. W wytycznych EFSA wskazuje się na ewentualną konieczność przeprowadzenia badań dodatkowych zależnie od cech rośliny zmodyfikowanej genetycznie oraz od wyników pierwszych badań. Zgodnie z opinią EFSA na ten temat 90-dniowe badania żywieniowe na gryzoniach karmionych całą żywnością lub paszą są, mimo swoich ograniczeń, w uzasadnionych warunkach podstawowym badaniem dodatkowym pozwalającym rozwiać wątpliwości, które pojawiły się w trakcie oceny ryzyka.
(11) Nie można jednak było z odpowiednią dokładnością zdefiniować poziomu wątpliwości, który wymagałby przedłożenia 90-dniowych badań żywieniowych. Ponadto niektóre organy państw członkowskich zajmujące się oceną żywności i paszy są zdania, że takie badanie należy przeprowadzać w przypadku wszystkich wniosków dotyczących roślin zmodyfikowanych genetycznie zawierających pojedyncze modyfikacje genetyczne. Z uwagi na wspomniane rozbieżne opinie, a także aby podnieść zaufanie konsumentów, badania takie powinny być obecnie wymagane w odniesieniu do wszystkich wniosków dotyczących roślin zmodyfikowanych genetycznie zawierających pojedyncze modyfikacje genetyczne oraz, w stosownych przypadkach, roślin zmodyfikowanych genetycznie zawierających złożone modyfikacje genetyczne.
(12) Badania mające na celu wykazanie, że genetycznie zmodyfikowana żywność lub pasza spełniają wymogi rozporządzenia (WE) nr 1829/2003, zakładające udział zwierząt laboratoryjnych, powinny być przeprowadzane zgodnie z dyrektywą Parlamentu Europejskiego i Rady 2010/63/UE z dnia 22 września 2010 r. w sprawie ochrony zwierząt wykorzystywanych do celów naukowych (7) i nie powinny wykraczać poza minimum niezbędne do odpowiedniego wykazania bezpieczeństwa genetycznie zmodyfikowanej żywności lub paszy. Obecne wątpliwości dotyczące konieczności przeprowadzania 90-dniowych badań żywieniowych i ich przebiegu będą przedmiotem dużego projektu badawczego przeprowadzanego w ramach tematu 2 programu prac na 2012 r. „Żywność, rolnictwo i rybołówstwo oraz biotechnologia” w siódmym programie ramowym w zakresie badań (7PR). Wymagania dotyczące badań karmienia zwierząt w kontekście oceny ryzyka stwarzanego przez organizmy zmodyfikowane genetycznie powinny zostać poddane przeglądowi w świetle wyników wspomnianego projektu; wyniki te powinny być dostępne najpóźniej do końca 2015 r. Pod uwagę należy także wziąć inne wiarygodne informacje naukowe, które mogą być dostępne w momencie składania wniosku.
(13) Przepisy ustanowione w niniejszym rozporządzeniu powinny obowiązywać w odniesieniu do wszystkich wniosków dotyczących roślin zmodyfikowanych genetycznie, natomiast rodzaj i konieczność przeprowadzania badań dotyczących oceny cech i bezpieczeństwa genetycznie zmodyfikowanej żywności lub paszy, będących przedmiotem wniosku, mogą się różnić, zależnie od natury modyfikacji genetycznej lub charakteru produktu. Na przykład modyfikacje genetyczne, które mają nieznaczny wpływ na skład genetycznie zmodyfikowanej żywności lub paszy, lub produkty wysoko przetworzone, co do których dowiedziono, że są identyczne z produktami wytwarzanymi z tradycyjnych odpowiedników, wymagają innych badań niż produkt będący wynikiem złożonych modyfikacji genetycznych mających na celu zmianę właściwości odżywczych.
(14) Wymagania ustanowione w niniejszym rozporządzeniu dotyczące badań, które należy włączyć do wniosku o zatwierdzenie zgodnie z rozporządzeniem (WE) nr 1829/2003, nie powinny stanowić dla EFSA przeszkody przed wymaganiem od wnioskodawcy, w stosownych przypadkach, uzupełnienia danych szczegółowych dołączonych do wniosku zgodnie z art. 6 ust. 2 i art. 18 ust. 2 rozporządzenia (WE) nr 1829/2003.
(15) Aby zagwarantować wysoką jakość badań i przejrzystość ich udokumentowania, konieczne jest, aby badania te były przeprowadzane zgodnie z odpowiednimi systemami zapewniania jakości, a we wszystkich przypadkach należy dołączać dane nieprzetworzone, podane w odpowiednim formacie elektronicznym. Badania toksykologiczne należy przeprowadzać zgodnie z zasadami zapewniania jakości określonymi w dyrektywie 2004/10/WE Parlamentu Europejskiego i Rady z dnia 11 lutego 2004 r. w sprawie harmonizacji przepisów ustawowych, wykonawczych i administracyjnych odnoszących się do stosowania zasad dobrej praktyki laboratoryjnej i weryfikacji jej stosowania na potrzeby badań substancji chemicznych (8). Jeżeli takie badania przeprowadzane są poza terytorium Unii, powinny być zgodne z zasadami dobrej praktyki laboratoryjnej OECD (GLP) odzwierciedlającymi aktualny stan wiedzy. Badania inne niż badania toksykologiczne powinny być przeprowadzane zgodnie z normami ISO lub standardami GLP.
(16) Należy również zdefiniować wymogi odnoszące się do składania dodatkowych informacji dotyczących bezpieczeństwa organizmów zmodyfikowanych genetycznie oraz piśmiennictwa naukowego dotyczącego ewentualnego wpływu produktów objętych wnioskiem na zdrowie i na środowisko.
(17) Podczas procesu modyfikacji genetycznej roślin i innych organizmów często stosuje się geny markerowe w celu ułatwienia selekcji i identyfikacji genetycznie zmodyfikowanych komórek, które wśród licznych komórek niepoddanych transformacji zawierają interesujące geny włączane do genomu organizmu gospodarza. Takie geny markerowe należy starannie selekcjonować. Ponadto obecnie możliwe jest tworzenie organizmów zmodyfikowanych genetycznie bez konieczności stosowania genów markerowych kodujących oporność na antybiotyki. W związku z powyższym, oraz zgodnie z art. 4 ust. 2 dyrektywy 2001/18/WE, wnioskodawca powinien starać się tworzyć organizmy zmodyfikowane genetycznie bez używania genów markerowych oporności na antybiotyki.
(18) W przeprowadzonym z zastosowaniem segregacji zbiorze genetycznie zmodyfikowanych roślin (segregacja upraw) zawierających złożone modyfikacje genetyczne znajdują się różne subkombinacje modyfikacji genetycznych. Obecne procedury kontrolne nie pozwalają ponadto na identyfikację źródła kombinacji modyfikacji genetycznych. W związku z powyższym, aby sprawić, by zezwolenia odpowiadały produktom, których wprowadzenie do obrotu jest nieuniknione, oraz w celu umożliwienia przeprowadzania kontroli, wnioski dotyczące genetycznie zmodyfikowanej żywności i paszy pochodzącej z segregowanych upraw powinny obejmować wszystkie jeszcze niezatwierdzone subkombinacje, niezależnie od ich źródła.
(19) Rozporządzenie (WE) nr 1829/2003 stanowi, że propozycja monitorowania stosowania genetycznie zmodyfikowanej żywności lub paszy po wprowadzeniu do obrotu powinna być składana przez wnioskodawcę tylko w stosownych przypadkach. Należy zatem wyszczególnić warunki, w których taka propozycja powinna – zgodnie z wynikiem oceny ryzyka – być dołączona do wniosku. Monitorowanie po wprowadzeniu do obrotu powinno być rozważane tylko w przypadkach, w których – niezależnie od wykazania bezpieczeństwa genetycznie zmodyfikowanej żywności lub paszy – właściwe jest potwierdzenie oczekiwanej konsumpcji, przestrzegania warunków stosowania lub zidentyfikowanych rezultatów. Na przykład, kiedy genetycznie zmodyfikowana żywność lub pasza zmieniły swój skład odżywczy albo kiedy wartości odżywcze takiej żywności lub paszy różnią się od wartości tradycyjnej żywności lub paszy, które miałyby zastąpić, albo kiedy istnieje prawdopodobieństwo zwiększonej alergenności z powodu modyfikacji genetycznej.
(20) Niniejsze rozporządzenie powinno uwzględniać zobowiązania Unii w zakresie handlu międzynarodowego oraz inne wymagania Protokołu z Kartageny o bezpieczeństwie biologicznym do Konwencji o różnorodności biologicznej, zatwierdzonego decyzją Rady 2002/628/WE z dnia 25 czerwca 2002 r. dotyczącą zawarcia w imieniu Wspólnoty Europejskiej Protokołu z Kartageny o bezpieczeństwie biologicznym do Konwencji o różnorodności biologicznej (9), a także przepisy rozporządzenia (WE) nr 1946/2003 Parlamentu Europejskiego i Rady z dnia 15 lipca 2003 r. w sprawie transgranicznego przemieszczania organizmów genetycznie zmodyfikowanych (10).
(21) Aby mieć pewność, że metody badań włączone do wniosku są właściwe do wykazania, że żywność lub pasza są zgodne z wymogami dotyczącymi zatwierdzania ustanowionymi w rozporządzeniu (WE) nr 1829/2003, badania te powinny być przeprowadzane zgodnie z niniejszym rozporządzeniem lub z wytycznymi uzgodnionymi na arenie międzynarodowej, np. z wytycznymi OECD, jeżeli takie istnieją. Aby w odniesieniu do metod badań zapewnić spełnienie tych samych norm przez wnioski o odnowienie, należy również stosować te wymogi do wniosków o odnowienie zatwierdzenia genetycznie zmodyfikowanej żywności i paszy.
(22) Aby zapewnić dokładne oznaczenie zmodyfikowanej genetycznie żywności lub paszy będących przedmiotem wniosku zgodnie z rozporządzeniem (WE) nr 1829/2003, wnioski powinny zawierać propozycję niepowtarzalnego identyfikatora dla każdego danego organizmu zmodyfikowanego genetycznie zgodnie z rozporządzeniem Komisji (WE) nr 65/2004 z dnia 14 stycznia 2004 r. ustanawiającym system ustanawiania oraz przypisywania niepowtarzalnych identyfikatorów organizmom zmodyfikowanym genetycznie (11).
(23) Niniejsze rozporządzenie zastępuje niektóre przepisy rozporządzenia (WE) nr 641/2004 w odniesieniu do genetycznie zmodyfikowanych roślin przeznaczonych na żywność lub paszę oraz żywności lub paszy zawierających rośliny zmodyfikowane genetycznie, składających się z nich lub z nich produkowanych. Niemniej jednak rozporządzenie (WE) nr 641/2004 powinno nadal obowiązywać w odniesieniu do pozostałych typów produktów zmodyfikowanych genetycznie, tj. genetycznie zmodyfikowanych zwierząt i mikroorganizmów. Ponadto niektóre przepisy tego rozporządzenia są nieaktualne. Należy zatem odpowiednio zmienić rozporządzenie (WE) nr 641/2004.
(24) Rozporządzenie Komisji (WE) nr 1981/2006 z dnia 22 grudnia 2006 r. ustalające szczegółowe zasady wykonania przepisów art. 32 rozporządzenia (WE) nr 1829/2003 Parlamentu Europejskiego i Rady w odniesieniu do wspólnotowego laboratorium referencyjnego ds. organizmów zmodyfikowanych genetycznie (12) należy zmienić w celu włączenia do niego odesłań do niniejszego rozporządzenia.
(25) Rozporządzenie (WE) nr 1829/2003 stanowi, że Komisja zasięga opinii EFSA przed określeniem przepisów wykonawczych odnoszących się do wniosków o zatwierdzenie na mocy tego rozporządzenia. Zasięgnięto opinii EFSA w odniesieniu do tych przepisów.
(26) Niniejsze rozporządzenie zostało sporządzone w oparciu o aktualną wiedzę naukową i techniczną. Komisja powinna monitorować rozwój sytuacji w tym zakresie oraz publikacje nowych lub dodatkowych wytycznych EFSA.
(27) Niniejsze rozporządzenie stosuje się do wniosków złożonych po jego wejściu w życie. Należy przewidzieć środki przejściowe w celu umożliwienia wnioskodawcom dostosowania się do tych przepisów oraz uniknięcia niepotrzebnych opóźnień w rozpatrywaniu złożonych wniosków lub wniosków przygotowanych już do złożenia.
(28) Środki przewidziane w niniejszym rozporządzeniu są zgodne z opinią Stałego Komitetu ds. Łańcucha Żywnościowego i Zdrowia Zwierząt,
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
ROZDZIAŁ I
PRZEPISY OGÓLNE
Artykuł 1
Zakres
Niniejsze rozporządzenie stosuje się do wniosków złożonych na podstawie art. 5, 11, 17 i 23 rozporządzenia (WE) nr 1829/2003 o zatwierdzenie:
a) genetycznie zmodyfikowanych roślin przeznaczonych na żywność lub paszę;
b) żywności lub paszy zawierających rośliny zmodyfikowane genetycznie lub składających się z nich;
c) żywności wyprodukowanej ze składników wyprodukowanych z roślin zmodyfikowanych genetycznie lub zawierających takie składniki, lub paszy wyprodukowanej z takich roślin.
Artykuł 2
Definicje
Do celów niniejszego rozporządzenia stosuje się definicje zawarte w rozporządzeniu (WE) nr 1829/2003.
Definicje „ryzyka” , „oceny ryzyka” i „zagrożenia” stosowane do celów niniejszego rozporządzenia są zgodne z definicjami zawartymi w art. 3 rozporządzenia (WE) nr 178/2002.
ROZDZIAŁ II
WYMAGANIA OGÓLNE
Artykuł 3
Sporządzanie i przedstawianie wniosków składanych na podstawie art. 5 ust. 1 i art. 17 ust. 1
1. Wniosek składany na podstawie art. 5 ust. 1 i art. 17 ust. 1 rozporządzenia (WE) nr 1829/2003:
a) jest składany zgodnie z wymaganiami dotyczącymi sporządzania i przedstawiania wniosków ustanowionymi w załączniku I;
b) zawiera wszystkie informacje wymagane na mocy załącznika I, zgodnie ze szczegółowymi wymaganiami art. 4, 5 i 6.
2. Wniosek zawiera, w odniesieniu do każdego szczegółowego wymagania ustanowionego w art. 4, 5 i 6:
a) streszczenia i wyniki badań, o których mowa we wniosku;
b) załączniki, w których znajdują się szczegółowe informacje dotyczące tych badań.
3. Wniosek zawiera listę kontrolą wykazującą, że informacje wymagane na mocy art. 4, 5 i 6 są kompletne.
4. Jeżeli wniosek jest ograniczony do zastosowania tylko jako żywność lub tylko jako pasza, zawiera możliwe do zweryfikowania uzasadnienie wyjaśniające, dlaczego zatwierdzenie nie obejmuje obu zastosowań zgodnie z art. 27 rozporządzenia (WE) nr 1829/2003.
5. W momencie składania wniosek zawiera jasne stwierdzenie, które części wniosku uznaje się za poufne, a także możliwe do zweryfikowania uzasadnienie zgodnie z art. 30 rozporządzenia (WE) nr 1829/2003.
Dodatkowe informacje składane w trakcie procedury zatwierdzania zawierają, w momencie ich składania, jasne stwierdzenie, które części tych dodatkowych informacji uznaje się za poufne, a także możliwe do zweryfikowania uzasadnienie zgodnie z art. 30 rozporządzenia (WE) nr 1829/2003.
6. W przypadku przedłożenia badań Europejskiemu Urzędowi ds. Bezpieczeństwa Żywności (EFSA) na potrzeby innego wniosku, a także, w stosownych przypadkach, w zakresie, w którym badania te mogą być użyte przez wnioskodawcę zgodnie z art. 31 rozporządzenia (WE) nr 1829/2003, odniesienia do tych badań oraz do wyników oceny EFSA mogą, za zgodą EFSA, zostać włączone do innego wniosku.
ROZDZIAŁ III
WYMAGANIA SZCZEGÓŁOWE
Artykuł 4
Wymagania dotyczące przeprowadzania badań związanych z wnioskami składanymi na podstawie art. 5 ust. 3 i art. 17 ust. 3
1. Badania toksykologiczne są przeprowadzane w ośrodkach, które są zgodne z:
a) wymogami dyrektywy 2004/10/WE; lub
b) „Zasadami dobrej praktyki laboratoryjnej OECD” (GLP), jeżeli badania te są przeprowadzane poza Unią.
Wnioskodawca dostarcza dowody wykazujące spełnienie powyższych wymagań.
2. Badania inne niż badania toksykologiczne:
a) są zgodne z zasadami dobrej praktyki laboratoryjnej (GLP) ustanowionymi w dyrektywie 2004/10/WE; lub
b) są przeprowadzane przez organizację akredytowaną na podstawie właściwej normy ISO.
3. Informacje dotyczące protokołów badań oraz wyników uzyskanych z badań, o których mowa w ust. 1 i 2, są wyczerpujące i zawierają dane nieprzetworzone, w formacie elektronicznym odpowiednim do przeprowadzania analizy statystycznej lub innej analizy.
Artykuł 5
Wymagania naukowe dotyczące oceny ryzyka stwarzanego przez genetycznie zmodyfikowaną żywność i paszę związanej z wnioskami składanymi na podstawie art. 5 ust. 3 i art. 17 ust. 3
1. Informacje, w tym badania, które należy dołączyć do wniosku, o których mowa w art. 5 ust. 3 lit. a)– f) i h) oraz w art. 17 ust. 3 lit. a)–f) i h) rozporządzenia (WE) nr 1829/2003, dostarcza się zgodnie z wymogami naukowymi dla oceny ryzyka stwarzanego przez genetycznie zmodyfikowaną żywność i paszę, ustanowionymi w załączniku II do niniejszego rozporządzenia.
2. W drodze odstępstwa od ust. 1 możliwe jest złożenie wniosku, który nie spełnia wszystkich wymagań tego ustępu, pod warunkiem że:
a) dana informacja nie jest konieczna ze względu na naturę modyfikacji genetycznej lub produktu; lub
b) dostarczenie takich informacji nie jest konieczne z naukowego punktu widzenia lub jest technicznie niemożliwe.
Wnioskodawca składa możliwe do zweryfikowania uzasadnienie odstępstwa.
3. Ustępy 1 i 2 nie stanowią dla EFSA przeszkody do wymagania od wnioskodawcy, w stosownych przypadkach, uzupełnienia danych szczegółowych dołączonych do wniosku zgodnie z art. 6 ust. 2 i art. 18 ust. 2 rozporządzenia (WE) nr 1829/2003.
Artykuł 6
Dodatkowe informacje dotyczące oceny ryzyka stwarzanego przez genetycznie zmodyfikowaną żywność i paszę związanej z wnioskami składanymi na podstawie art. 5 ust. 3 i art. 17 ust. 3
1. Oprócz informacji wymaganych zgodnie z art. 5 i załącznikiem II wniosek zawiera systematyczny przegląd badań opublikowanych w literaturze naukowej i badań wykonanych przez wnioskodawcę w okresie 10 lat przed złożeniem dokumentacji dotyczącej ewentualnego wpływu genetycznie zmodyfikowanej żywności i paszy objętej wnioskiem na zdrowie ludzi i zwierząt.
2. W trakcie procedury zatwierdzania wnioskodawca bez zbędnej zwłoki przekazuje EFSA dodatkowe informacje, które mogłyby wpłynąć na ocenę ryzyka stwarzanego przez genetycznie zmodyfikowaną żywność lub paszę po złożeniu wniosku. Wnioskodawca w szczególności przekazuje EFSA informacje dotyczące zakazów lub ograniczeń, nałożonych przez właściwy organ dowolnego państwa trzeciego na podstawie oceny ryzyka stwarzanego przez genetycznie zmodyfikowaną żywność lub paszę.
Artykuł 7
Wymagania stosowane do monitorowania genetycznie zmodyfikowanej żywności lub paszy po wprowadzeniu do obrotu, związanego z wnioskami składanymi na podstawie art. 5 ust. 3 i art. 17 ust. 3
1. Wnioskodawca składa propozycję monitorowania po wprowadzeniu do obrotu w odniesieniu do stosowania żywności i paszy zgodnie z art. 5 ust. 3 lit. k) oraz art. 17 ust. 3 lit. k) rozporządzenia (WE) nr 1829/2003, jeżeli informacje przedstawione zgodnie z art. 4, 5 i 6 wykazują, że genetycznie zmodyfikowana żywność i pasza są zgodne z art. 4 ust. 1 i art. 16 ust. 1 rozporządzenia (WE) nr 1829/2003, oraz jeżeli, zgodnie z wynikiem oceny ryzyka, należy potwierdzić:
a) że konsumenci/właściciele zwierząt przestrzegają specjalnych zaleceń dotyczących stosowania;
b) przewidywaną konsumpcję genetycznie zmodyfikowanej żywności lub paszy; lub
c) znaczenie i intensywność skutków i niezamierzonych skutków wykrytych podczas oceny ryzyka przed wprowadzeniem do obrotu, które mogą być określone jedynie przez monitorowanie po wprowadzeniu do obrotu.
2. Wnioskodawca dopilnowuje, aby monitorowanie po wprowadzeniu do obrotu:
a) odbywało się w celu zebrania wiarygodnych informacji w odniesieniu do jednego lub kilku aspektów wymienionych w ust. 1. Informacje te pozwalają na wykrycie, czy ewentualny wpływ na zdrowie lub niepożądane skutki dla zdrowia można powiązać z konsumpcją genetycznie zmodyfikowanej żywności lub paszy;
b) było oparte na strategiach zmierzających do zebrania właściwych informacji od konkretnych zainteresowanych stron, w tym konsumentów, oraz na wiarygodnym i potwierdzonym przepływie informacji między różnymi zainteresowanymi stronami. Bardziej szczegółowe strategie są stosowane w przypadkach, kiedy konieczne jest zebranie danych dotyczących indywidualnego spożycia konkretnej żywności lub spożycia wśród konkretnej grupy wiekowej;
c) było odpowiednio uzasadnione i wiązało się z dogłębnym opisem wybranych metodologii dla proponowanego monitorowania po wprowadzeniu do obrotu, a także obejmowało aspekty związane z analizą zebranych informacji.
Artykuł 8
Wymagania dotyczące metod wykrywania, identyfikacji i kwantyfikacji oraz pobierania próbek kontrolnych i materiału odniesienia genetycznie zmodyfikowanej żywności lub paszy, związanych z wnioskami składanymi na podstawie art. 5 ust. 3, art. 11 ust. 2, art. 17 ust. 3 i art. 23 ust. 2
1. Wnioski składane na podstawie art. 5 ust. 1 i art. 17 ust. 1 rozporządzenia (WE) nr 1829/2003 są zgodne z następującymi wymaganiami, o których mowa w art. 5 ust. 3 lit. i) i j) oraz art. 17 ust. 3 lit. i) i j) tego rozporządzenia, wyszczególnionymi w załączniku III do niniejszego rozporządzenia, w odniesieniu do:
a) metod wykrywania i identyfikacji modyfikacji genetycznych;
b) próbek żywności lub paszy oraz ich próbek kontrolnych, a także informacji dotyczących miejsca, w którym dostępny jest materiał odniesienia.
2. W odniesieniu do wniosków składanych na podstawie art. 11 ust. 1 i art. 23 ust. 1 rozporządzenia (WE) nr 1829/2003 wymagania wyszczególnione w załączniku III do niniejszego rozporządzenia w odniesieniu do:
a) metod wykrywania i identyfikacji modyfikacji genetycznych;
b) próbek żywności lub paszy oraz ich próbek kontrolnych, a także informacji dotyczących miejsca, w którym dostępny jest materiał odniesienia;
stosuje się jedynie na potrzeby stosowania art. 11 ust. 2 lit. d) i art. 23 ust. 2 lit. d).
ROZDZIAŁ IV
PRZEPISY PRZEJŚCIOWE I KOŃCOWE
Artykuł 9
Przepisy przejściowe
1. Do dnia 8 grudnia 2013 r. wnioskodawcy mogą składać wnioski objęte zakresem niniejszego rozporządzenia na podstawie rozporządzenia (WE) nr 641/2004 w wersji tego rozporządzenia obowiązującej w dniu 8 czerwca 2013 r.
2. W drodze odstępstwa od art. 4 ust. 2, w przypadku badań rozpoczętych przed dniem wejścia w życie niniejszego rozporządzenia oraz przeprowadzanych zgodnie z systemami zapewniania jakości innymi niż GLP i ISO, wnioskodawca dostarcza:
a) szczegółowy opis systemu zapewniania jakości, zgodnie z którym te badania są przeprowadzane; oraz
b) wyczerpujące informacje dotyczące protokołów badań oraz wyników uzyskanych z badań, w tym dane nieprzetworzone.
Artykuł 10
Zmiany w rozporządzeniu (WE) nr 641/2004
W rozporządzeniu (WE) nr 641/2004 wprowadza się następujące zmiany:
1) artykuł 1 otrzymuje brzmienie:
„Artykuł 1
Niniejszy rozdział przewiduje szczegółowe zasady dotyczące wniosków o zatwierdzenie, przedkładanych zgodnie z art. 5 i 17 rozporządzenia (WE) nr 1829/2003, z wyjątkiem wniosków objętych rozporządzeniem wykonawczym Komisji (UE) nr 503/2013 (*)
___ ______
(*) Dz.U. L 157 z 8.6.2013, s. 1.”;
2) skreśla się art. 5–19.
Artykuł 11
Zmiany w rozporządzeniu (WE) nr 1981/2006
W rozporządzeniu (WE) nr 1981/2006 wprowadza się następujące zmiany:
1) art. 2 lit. a) otrzymuje brzmienie:
„a) »pełna procedura uwierzytelnienia« oznacza:
(i) ocenę – przeprowadzaną w drodze badań międzylaboratoryjnych z udziałem krajowych laboratoriów referencyjnych – kryteriów skuteczności metod przyjętych przez wnioskodawcę jako zgodnych z dokumentem »Definicja minimalnych wymogów działania dotyczących analitycznych metod testowania OGZ«, o którym mowa w:
– w przypadku roślin zmodyfikowanych genetycznie przeznaczonych na żywność lub paszę, żywności lub paszy zawierających rośliny zmodyfikowane genetycznie lub składających się z nich oraz żywności produkowanej ze składników wyprodukowanych z roślin zmodyfikowanych genetycznie lub zawierającej takie składniki, lub paszy produkowanej z roślin zmodyfikowanych genetycznie – w pkt 3.1.C.4 załącznika III do rozporządzenia wykonawczego Komisji (UE) nr 503/2013 (*);
– we wszystkich innych przypadkach – w pkt 1(B) załącznika I do rozporządzenia (WE) nr 641/2004;
oraz
(ii) ocenę precyzji i poprawności metody dostarczonej przez wnioskodawcę.
_________
(*) Dz.U. L 157 z 8.6.2013, s. 1.”;
2) art. 3 ust. 2 akapit pierwszy i drugi otrzymuje brzmienie:
„2. Na żądanie wspólnotowego laboratorium referencyjnego wnioskodawca wnosi dodatkową opłatę w wysokości 60 000 EUR, jeżeli konieczne jest przeprowadzenie pełnej procedury uwierzytelnienia metody wykrywania i identyfikacji pojedynczej modyfikacji genetycznej zgodnie z wymaganiami ustanowionymi w następujących przepisach:
a) załącznik III do rozporządzenia wykonawczego (UE) nr 503/2013, jeżeli wniosek dotyczy:
(i) genetycznie zmodyfikowanych roślin przeznaczonych na żywność lub paszę;
(ii) żywności lub paszy zawierających rośliny zmodyfikowane genetycznie lub składających się z nich;
(iii) żywności produkowanej ze składników wyprodukowanych z roślin zmodyfikowanych genetycznie lub zawierających takie składniki, lub paszy wyprodukowanej z takich roślin; lub
b) we wszystkich innych przypadkach – pkt 1(B) załącznika I do rozporządzenia (WE) nr 641/2004.
Stawkę tę należy przemnożyć przez liczbę modyfikacji genetycznych podlegających pełnemu uwierzytelnieniu.”.
Artykuł 12
Przegląd
1. Komisja monitoruje stosowanie niniejszego rozporządzenia, rozwój wiedzy naukowej w zakresie ograniczenia korzystania ze zwierząt w procedurach naukowych, rezygnacji z ich stosowania na rzecz innych metod oraz poprawy warunków korzystania z nich, a także publikacje nowych wytycznych EFSA. Komisja monitoruje w szczególności wyniki projektu badawczego o nazwie GRACE („ GMO Risk Assessment and Communication of Evidence”) prowadzonego w ramach programu prac na 2012 r. w siódmym programie ramowym w zakresie badań (7PR).
2. Komisja poddaje przeglądowi wymóg przeprowadzenia 90-dniowych badań żywieniowych na gryzoniach karmionych całą genetycznie zmodyfikowaną żywnością lub paszą (pkt 1.4.4.1 załącznika II) na podstawie nowych danych naukowych. Wyniki tego przeglądu są publikowane najpóźniej do dnia 30 czerwca 2016 r.
Artykuł 13
Wejście w życie
Niniejsze rozporządzenie wchodzi w życie dwudziestego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich
Sporządzono w Brukseli dnia 3 kwietnia 2013 r.
|
| W imieniu Komisji |
| José Manuel BARROSO | |
| Przewodniczący |
(1) Dz.U. L 268 z 18.10.2003, s. 1.
(2) Dz.U. L 31 z 1.2.2002, s. 1.
(3) Dz.U. L 102 z 7.4.2004, s. 14.
(4) Dz.U. L 106 z 17.4.2001, s. 1.
(5) Komisja Kodeksu Żywnościowego, GL 45-2003.
(6) Dziennik EFSA 2011; 9(5):2150.
(7) Dz.U. L 276 z 20.10.2010, s. 33.
(8) Dz.U. L 50 z 20.2.2004, s. 44.
(9) Dz.U. L 201 z 31.7.2002, s. 48.
(10) Dz.U. L 287 z 5.11.2003, s. 1.
(11) Dz.U. L 10 z 14.1.2004, s. 5.
(12) Dz.U. L 368 z 23.12.2006, s. 99.
ZAŁĄCZNIK I
SPORZĄDZANIE I PRZEDKŁADANIE WNIOSKÓW
Wniosek zawiera następujące informacje:
CZĘŚĆ I
INFORMACJE OGÓLNE
1. Nazwa i adres wnioskodawcy (przedsiębiorstwo lub instytut).
2. Nazwisko, kwalifikacje i doświadczenie naukowców odpowiedzialnych i dane kontaktowe osoby odpowiedzialnej za wszelkie kontakty z Europejskim Urzędem ds. Bezpieczeństwa Żywności (EFSA).
3. Opis i specyfikacja rośliny zmodyfikowanej genetycznie i jej produktów.
4. Zakres wniosku
a) genetycznie zmodyfikowana żywność
☐ żywność zawierająca rośliny zmodyfikowane genetycznie lub składająca się z takich roślin
☐ żywność wyprodukowana z roślin zmodyfikowanych genetycznie lub zawierająca składniki wyprodukowane z roślin zmodyfikowanych genetycznie
b) genetycznie zmodyfikowana pasza
☐ pasza zawierająca rośliny zmodyfikowane genetycznie lub składająca się z takich roślin
☐ pasza wyprodukowana z roślin zmodyfikowanych genetycznie
c) genetycznie zmodyfikowane rośliny przeznaczone na żywność lub paszę
☐ produkty inne niż żywność i pasza, zawierające rośliny zmodyfikowane genetycznie lub składające się z takich roślin, z wyjątkiem uprawy
☐ nasiona i inny materiał rozmnożeniowy roślin do uprawy w Unii.
5. Niepowtarzalny identyfikator
Propozycja niepowtarzalnego identyfikatora dla rośliny zmodyfikowanej genetycznie, opracowana zgodnie z rozporządzeniem (WE) nr 65/2004.
6. W stosownych przypadkach – szczegółowy opis metody produkcji i wytwarzania.
Opis ten może obejmować na przykład szczegółowy opis specyficznych metod produkcji żywności lub paszy, wynikających z charakteru modyfikacji genetycznej lub prowadzących do wytworzenia żywności lub paszy o określonych cechach.
7. W stosownych przypadkach – warunki wprowadzania do obrotu genetycznie zmodyfikowanej żywności lub paszy, w tym warunki szczególne dotyczące obchodzenia się z nimi i ich stosowania.
8. W stosownych przypadkach – status żywności lub paszy, lub powiązanych substancji na podstawie innych przepisów prawa unijnego.
Dodatkowe wymogi dotyczące zezwolenia przewidziane w prawie unijnym, związane z wprowadzaniem do obrotu żywności lub paszy lub z mającym zastosowanie „maksymalnym poziomem pozostałości”, w przypadku gdy żywność lub pasza mogą zawierać pozostałości środków ochrony roślin.
CZĘŚĆ II
INFORMACJE NAUKOWE
We wniosku uwzględnia się wszystkie wymagania zawarte w części II, chyba że takie wymagania nie są uzasadnione zakresem wniosku (na przykład w przypadku gdy wniosek jest ograniczony do żywności lub paszy produkowanej z GMO).
1. IDENTYFIKACJA I CHARAKTERYSTYKA ZAGROŻEŃ
1.1. Informacje dotyczące roślin biorców lub (w stosownych przypadkach) roślin rodzicielskich
a) pełna nazwa:
(i) rodzina;
(ii) rodzaj;
(iii) gatunek;
(iv) podgatunek;
(v) kultywar, linia hodowlana;
(vi) nazwa zwyczajowa;
b) geograficzne rozmieszczenie i uprawa rośliny w obrębie Unii;
c) informacje na temat roślin biorców lub rodzicielskich istotne dla ich bezpieczeństwa, w tym na temat znanej toksyczności lub alergenności;
d) dane dotyczące przeszłego i obecnego zastosowania rośliny biorcy, np. historii bezpiecznego stosowania jako żywności lub paszy przeznaczonych do spożycia, w tym informacje na temat zwykłych sposobów uprawiania, transportowania i przechowywania danej rośliny, tego, czy potrzebne jest specjalne przetwarzanie, aby można było bezpiecznie spożywać daną roślinę, normalnej roli danej rośliny w diecie (np. która część rośliny jest wykorzystywana jako źródło żywności, czy jej spożycie jest ważne w szczególnych podgrupach populacji, jakie ważne makro- lub mikroskładniki pokarmowe wnosi do diety);
e) dodatkowe informacje dotyczące roślin biorców lub rodzicielskich wymagane w odniesieniu do aspektów bezpieczeństwa dla środowiska:
(i) informacje dotyczące rozmnażania:
– sposoby rozmnażania,;
– szczególne czynniki wpływające na rozmnażanie (jeżeli takie występują),
– czas trwania pokolenia;
(ii) zgodność płciowa z innymi gatunkami roślin uprawnych lub dzikich;
(iii) zdolność przetrwania:
– zdolność do tworzenia form przetrwalnikowych,
– szczególne czynniki wpływające na zdolność przetrwania, jeżeli takie występują;
(iv) rozprzestrzenianie:
– sposoby i zasięg rozprzestrzeniania (uwzględnić np. szacunkową ocenę zmniejszania się stężenia zdolnych do życia pyłków lub nasion w miarę zwiększania się odległości od roślin),
– szczególne czynniki wpływające na rozprzestrzenianie, jeżeli takie istnieją;
(v) geograficzne rozmieszczenie w obrębie Unii gatunków zgodnych płciowo;
(vi) w przypadku gatunków roślin, które nie występują w Unii, opis naturalnego siedliska roślin, obejmujący informacje na temat naturalnych drapieżników, pasożytów, konkurentów i symbiontów;
(vii) inne potencjalne wzajemne oddziaływania między rośliną zmodyfikowaną genetycznie a organizmami w ekosystemie, w którym zwykle ona występuje lub jest wykorzystywana w innych miejscach, z uwzględnieniem toksycznego wpływu na ludzi, zwierzęta i inne organizmy.
1.2. Charakterystyka molekularna
1.2.1. Informacje dotyczące modyfikacji genetycznej
1.2.1.1. Opis metod zastosowanych w celu dokonania modyfikacji genetycznej
1.2.1.2. Rodzaj i źródło wykorzystywanego wektora
1.2.1.3. Źródło kwasu(-ów) nukleinowego(-ych) dawcy wykorzystywanego(-ych) dla celów transformacji, wielkość i zamierzona funkcja każdego fragmentu stanowiącego część składową regionu przeznaczonego do insercji
1.2.2. Informacje o roślinie zmodyfikowanej genetycznie
1.2.2.1. Ogólny opis cech i właściwości, które zostały wprowadzone lub zmienione
1.2.2.2. Informacje dotyczące sekwencji, które zostały faktycznie dodane/usunięte
1.2.2.3. Informacje dotyczące ekspresji insertu
1.2.2.4. Stabilność genetyczna insertu i stabilność fenotypowa rośliny zmodyfikowanej genetycznie
1.2.2.5. Potencjalne ryzyko związane z poziomym transferem genów
1.2.3. Dodatkowe informacje dotyczące rośliny zmodyfikowanej genetycznie wymagane w odniesieniu do aspektów bezpieczeństwa dla środowiska
1.2.3.1. Informacje dotyczące różnic między rośliną zmodyfikowaną genetycznie a rośliną biorcą pod względem rozmnażania, rozprzestrzeniania, zdolności przetrwania lub innych właściwości
1.2.3.2. Jakiekolwiek zmiany zachodzące w roślinie zmodyfikowanej genetycznie dotyczące zdolności do transferu materiału genetycznego do innych organizmów, a mianowicie:
a) transfer genów z rośliny do bakterii;
b) transfer genów z rośliny do rośliny.
1.2.4. Wnioski z charakterystyki molekularnej
1.3. Analiza porównawcza
1.3.1. Wybór tradycyjnego odpowiednika i dodatkowych okazów porównawczych
1.3.2. Schemat doświadczenia i statystyczna analiza danych z doświadczeń polowych na potrzeby analizy porównawczej
1.3.2.1. Opis protokołów schematu doświadczenia
1.3.2.2. Analiza statystyczna
1.3.3. Wybór materiału i związków do analizy
1.3.4. Analiza porównawcza składu
1.3.5. Analiza porównawcza właściwości agronomicznych i fenotypowych
1.3.6. Skutki przetwarzania
1.3.7. Wniosek
1.4. Toksykologia
1.4.1. Badanie nowych białek ulegających ekspresji
1.4.2. Badanie nowych składników innych niż białka
1.4.3. Informacje na temat naturalnych składników żywności i paszy
1.4.4. Badanie całej genetycznie zmodyfikowanej żywności lub paszy
1.4.4.1. 90-dniowe badania żywieniowe na gryzoniach
1.4.4.2. Badania na zwierzętach w odniesieniu do szkodliwego wpływu na rozrodczość oraz do toksyczności rozwojowej lub przewlekłej
1.4.4.3. Inne badania na zwierzętach w celu ocenienia bezpieczeństwa i cech genetycznie zmodyfikowanej żywności i paszy
1.4.5. Wnioski z oceny toksykologicznej
1.5. Alergenność
1.5.1. Ocena alergenności nowych białek ulegających ekspresji
1.5.2. Ocena alergenności całej rośliny zmodyfikowanej genetycznie
1.5.3. Wniosek z oceny alergenności
1.6. Ocena wartości odżywczej
1.6.1. Ocena wartości odżywczych genetycznie zmodyfikowanej żywności
1.6.2. Ocena wartości odżywczych genetycznie zmodyfikowanej paszy
1.6.3. Wniosek z oceny wartości odżywczej
2. OCENA NARAŻENIA – PRZEWIDYWANE SPOŻYCIE LUB ZAKRES ZASTOSOWANIA
3. CHARAKTERYSTYKA RYZYKA
4. MONITOROWANIE GENETYCZNIE ZMODYFIKOWANEJ ŻYWNOŚCI LUB PASZY PO WPROWADZENIU DO OBROTU
5. OCENA ODDZIAŁYWANIA NA ŚRODOWISKO
6. PLAN MONITOROWANIA SKUTKÓW DLA ŚRODOWISKA
7. DODATKOWE INFORMACJE DOTYCZĄCE BEZPIECZEŃSTWA GENETYCZNIE ZMODYFIKOWANEJ ŻYWNOŚCI LUB PASZY
Częścią wniosku jest systematyczny przegląd badań opublikowanych w literaturze naukowej i badań wykonanych przez wnioskodawcę w okresie 10 lat przed złożeniem dokumentacji dotyczącej ewentualnego wpływu genetycznie zmodyfikowanej żywności i paszy objętej wnioskiem na zdrowie ludzi i zwierząt. Ten systematyczny przegląd przeprowadza się przy uwzględnieniu wytycznych EFSA dotyczących stosowania metod systematycznego przeglądu do ocen bezpieczeństwa żywności i paszy na potrzeby wspierania procesów decyzyjnych (1).
Jeżeli informacje uzyskane z tych badań nie są spójne z informacjami uzyskanymi z badań przeprowadzonych zgodnie z wymogami określonymi w załączniku II, wnioskodawca dostarcza szczegółową analizę odpowiednich badań i przedstawia wiarygodne wyjaśnienie zaobserwowanych różnic.
Wnioskodawca dostarcza dodatkowe informacje, które mogą mieć wpływ na ocenę bezpieczeństwa genetycznie zmodyfikowanej żywności lub paszy opracowaną w następstwie złożenia wniosku, jak również wszelkie informacje dotyczące zakazów lub ograniczeń nałożonych przez właściwy organ państwa trzeciego na podstawie oceny bezpieczeństwa.
CZĘŚĆ III
PROTOKÓŁ Z KARTAGENY
Wniosek zawiera informacje wymagane na podstawie art. 5 ust. 3 lit. c) i art. 17 ust. 3 lit. c) rozporządzenia (WE) nr 1829/2003 w celu spełnienia wymagań załącznika II do Protokołu z Kartageny o bezpieczeństwie biologicznym do Konwencji o różnorodności biologicznej.
Dostarczane informacje zawierają jako minimum informacje określone w załączniku II do rozporządzenia (WE) nr 1946/2003 Parlamentu Europejskiego i Rady (2):
a) nazwę oraz dane kontaktowe podmiotu składającego wniosek o podjęcie decyzji w sprawie stosowania krajowego;
b) nazwę oraz dane kontaktowe organu odpowiedzialnego za podjęcie decyzji;
c) nazwę oraz tożsamość GMO;
d) opis modyfikacji genetycznej, wykorzystanej techniki oraz wynikające z tego właściwości GMO;
e) każdą niepowtarzalną identyfikację GMO;
f) status taksonomiczny, nazwę zwyczajową, punkt zbierania lub uzyskiwania oraz właściwości organizmu biorcy lub organizmu rodzicielskiego związane z bezpieczeństwem biologicznym;
g) centra pochodzenia oraz centra różnorodności biologicznej, jeżeli są znane, organizmu biorcy lub organizmu rodzicielskiego oraz opis siedlisk, w których organizmy mogą utrzymywać się przy życiu lub rozmnażać się;
h) status taksonomiczny, nazwę zwyczajową, punkt zbierania lub uzyskiwania oraz właściwości organizmu dawcy lub organizmów dawców związane z bezpieczeństwem biologicznym;
i) zatwierdzone zastosowania GMO;
j) sprawozdanie z oceny ryzyka zgodnie z załącznikiem II do dyrektywy 2001/18/WE;
k) proponowane metody bezpiecznego obchodzenia się z GMO, jego składowania, transportu oraz stosowania, w tym pakowanie, etykietowanie, dokumentacja, a także, w stosownych przypadkach, unieszkodliwianie oraz procedury awaryjne.
CZĘŚĆ IV
ETYKIETOWANIE
Wniosek zawiera:
a) propozycję etykietowania we wszystkich językach urzędowych Unii, jeżeli propozycja określonego etykietowania wymagana jest zgodnie z art. 5 ust. 3 lit. f) oraz art. 17 ust. 3 lit. f) rozporządzenia (WE) nr 1829/2003;
b) uzasadnione oświadczenie stwierdzające, że żywność lub pasza nie budzą zastrzeżeń natury etycznej lub religijnej, albo propozycję etykietowania we wszystkich językach urzędowych Unii zgodnie z art. 5 ust. 3 lit. g) oraz art. 17 ust. 3 lit. g) rozporządzenia (WE) nr 1829/2003;
c) w stosownych przypadkach – propozycję etykietowania spełniającego wymogi części A pkt 8 załącznika IV do dyrektywy 2001/18/WE.
CZĘŚĆ V
METODY WYKRYWANIA, POBIERANIA PRÓBEK I IDENTYFIKACJI ORAZ MATERIAŁ ODNIESIENIA
Wnioskodawca przedstawia laboratorium referencyjnemu Unii Europejskiej (EURL), o którym mowa w art. 32 rozporządzenia (WE) 1829/2003, metody wykrywania, pobierania próbek i identyfikacji, jak również próbki żywności lub paszy oraz ich próbki kontrolne.
Wniosek zawiera kopię wypełnionego formularza złożenia wspomnianych próbek do EURL i dowód ich wysłania do EURL.
Wniosek zawiera informacje dotyczące miejsca, w którym dostępny jest materiał odniesienia.
Wnioskodawca postępuje zgodnie z instrukcjami dotyczącymi przygotowania i wysyłania próbek, zapewnionymi przez laboratorium referencyjne UE (EURL), o którym mowa w art. 32 rozporządzenia (WE) 1829/2003. Instrukcje te publikowane są na następującej stronie internetowej: http://gmo-crl.jrc.ec.europa.eu/guidancedocs.htm.
CZĘŚĆ VI
DODATKOWE INFORMACJE, KTÓRE NALEŻY DOSTARCZYĆ W ODNIESIENIU DO ROŚLIN ZMODYFIKOWANYCH GENETYCZNIE BĄDŹ ŻYWNOŚCI LUB PASZY ZAWIERAJĄCYCH ROŚLINY ZMODYFIKOWANE GENETYCZNIE LUB SKŁADAJĄCYCH SIĘ Z TAKICH ROŚLIN
Informacje wymagane w zgłoszeniu zgodnie z załącznikiem III do dyrektywy 2001/18/WE dostarcza się, jeżeli nie są objęte wymogami zawartymi w innych częściach wniosku.
CZĘŚĆ VII
STRESZCZENIE WNIOSKU
W niniejszej części określa się znormalizowaną formę, z którą streszczenie dokumentacji wniosku musi być zgodne.
W zależności od zakresu wniosku niektóre z wymaganych informacji mogą nie mieć zastosowania.
Streszczenie nie zawiera części uznawanych za poufne zgodnie z art. 30 rozporządzenia (WE) nr 1829/2003.
1. INFORMACJE OGÓLNE
1.1. Szczegóły wniosku
a) państwo członkowskie, którego dotyczy wniosek
b) numer wniosku
c) nazwa produktu (nazwy handlowe i wszelkie inne nazwy)
d) data potwierdzenia odbioru ważnego wniosku
1.2. Wnioskodawca
a) nazwa/imię i nazwisko wnioskodawcy
b) adres wnioskodawcy
c) nazwa/imię i nazwisko oraz adres przedstawiciela wnioskodawcy, mającego siedzibę w Unii (jeżeli wnioskodawca nie ma siedziby w Unii)
1.3. Zakres wniosku
a) genetycznie zmodyfikowana żywność
☐ żywność zawierająca rośliny zmodyfikowane genetycznie lub składająca się z takich roślin
☐ żywność wyprodukowana z roślin zmodyfikowanych genetycznie lub zawierająca składniki wyprodukowane z roślin zmodyfikowanych genetycznie
b) genetycznie zmodyfikowana pasza
☐ pasza zawierająca rośliny zmodyfikowane genetycznie lub składająca się z takich roślin
☐ pasza wyprodukowana z roślin zmodyfikowanych genetycznie
c) genetycznie zmodyfikowane rośliny przeznaczone na żywność lub paszę
☐ produkty inne niż żywność i pasza, zawierające rośliny zmodyfikowane genetycznie lub składające się z takich roślin, z wyjątkiem uprawy
☐ nasiona i materiał rozmnożeniowy roślin do uprawy w Unii
1.4. Czy produkt lub zastosowania powiązanych środków ochrony roślin są już zatwierdzone lub objęte inną procedurą udzielania zezwolenia w Unii?
Nie ☐
Tak ☐ (w tym przypadku należy określić)
1.5. Czy roślina zmodyfikowana genetycznie została zgłoszona na podstawie części B dyrektywy 2001/18/WE?
Tak ☐
Nie ☐ (w tym przypadku należy przedstawić dane dotyczące analizy ryzyka na podstawie elementów części B dyrektywy 2001/18/WE)
1.6. Czy roślina zmodyfikowana genetycznie lub produkty pochodne zostały wcześniej zgłoszone do obrotu w Unii na podstawie części C dyrektywy 2001/18/WE?
Nie ☐
Tak ☐ (w tym przypadku należy określić)
1.7. Czy produkt był przedmiotem wniosku lub został zatwierdzony w państwie trzecim, wcześniej lub równocześnie w stosunku do niniejszego wniosku?
Nie ☐
Tak ☐ W takim przypadku należy podać państwo trzecie, datę złożenia wniosku i, w miarę możliwości, przedstawić kopię wniosków z oceny ryzyka, podać datę wydania zezwolenia oraz zakres wniosku
1.8. Ogólny opis produktu
a) nazwa rośliny biorcy lub rośliny rodzicielskiej i zamierzona funkcja modyfikacji genetycznej
b) rodzaje produktów, które planuje się wprowadzić do obrotu zgodnie z zezwoleniem będącym przedmiotem wniosku, i wszelkie postaci, w których produkt nie może być wprowadzony do obrotu (np. nasiona, kwiaty cięte, części wegetatywne), w odniesieniu do których ograniczenia te proponowane są jako warunek zezwolenia będącego przedmiotem wniosku
c) zamierzone zastosowanie produktu i rodzaje użytkowników
d) wszelkie szczegółowe instrukcje i zalecenia dotyczące stosowania i przechowywania produktu oraz obchodzenia się z nim, w tym obowiązkowe ograniczenia proponowane jako warunek zezwolenia będącego przedmiotem wniosku
e) w stosownych przypadkach – obszary geograficzne w obrębie Unii, do których produkt ma się ograniczać zgodnie z warunkami zezwolenia będącego przedmiotem wniosku
f) każdy rodzaj środowiska, dla którego produkt jest nieodpowiedni
g) wszelkie proponowane wymogi dotyczące pakowania
h) wszelkie proponowane wymogi dotyczące etykietowania oprócz tych wymaganych na podstawie obowiązującego prawodawstwa UE innego niż rozporządzenie (WE) nr 1829/2003 oraz, w razie potrzeby, propozycja określonego etykietowania zgodnie z art. 13 ust. 2 i 3, art. 25 ust. 2 lit. c) i d) oraz art. 25 ust. 3 rozporządzenia (WE) nr 1829/2003.
W przypadku produktów innych niż żywność i pasza zawierających rośliny zmodyfikowane genetycznie lub składających się z nich należy podać propozycję etykietowania spełniającego wymogi części A pkt 8 załącznika IV do dyrektywy 2001/18/WE
i) szacowane potencjalne zapotrzebowanie
(i) w UE
(ii) na eksportowych rynkach UE
j) niepowtarzalny identyfikator zgodnie z rozporządzeniem (WE) nr 65/2004.
1.9. Sugerowane przez wnioskodawcę środki, jakie należy zastosować w przypadku niezamierzonego uwolnienia lub niewłaściwego zastosowania produktu, oraz środki służące unieszkodliwianiu i przetwarzaniu produktu
2. INFORMACJE DOTYCZĄCE ROŚLIN BIORCÓW LUB (W STOSOWNYCH PRZYPADKACH) ROŚLIN RODZICIELSKICH
2.1. Pełna nazwa
a) rodzina
b) rodzaj
c) gatunek
d) podgatunek
e) kultywar/linia hodowlana
f) nazwa zwyczajowa
2.2. Geograficzne rozmieszczenie i uprawa rośliny, w tym rozmieszczenie w obrębie Unii
2.3. Informacje dotycząca rozmnażania (w odniesieniu do aspektów bezpieczeństwa dla środowiska)
a) sposoby rozmnażania
b) szczególne czynniki wpływające na rozmnażanie
c) czas trwania pokolenia
2.4. Zgodność płciowa z innymi gatunkami roślin uprawnych lub dzikich (w odniesieniu do aspektów bezpieczeństwa dla środowiska)
2.5. Zdolność przetrwania (w odniesieniu do aspektów bezpieczeństwa dla środowiska)
a) zdolność do tworzenia form przetrwalnikowych
b) szczególne czynniki wpływające na zdolność przetrwania
2.6. Rozprzestrzenianie (w odniesieniu do aspektów bezpieczeństwa dla środowiska)
a) sposoby i zasięg rozprzestrzeniania
b) szczególne czynniki wpływające na rozprzestrzenianie
2.7. Geograficzne rozmieszczenie w obrębie Unii gatunków zgodnych płciowo (w odniesieniu do aspektów bezpieczeństwa dla środowiska)
2.8. W przypadku gatunków roślin, które w zwykłych warunkach nie występują w Unii, opis naturalnego siedliska roślin, obejmujący informacje na temat naturalnych drapieżników, pasożytów, konkurentów i symbiontów (w odniesieniu do aspektów bezpieczeństwa dla środowiska)
2.9. Inne potencjalne wzajemne oddziaływania, istotne dla tej rośliny zmodyfikowanej genetycznie, między tą rośliną a organizmami w ekosystemie, w którym zwykle ona występuje, lub w innym miejscu, w którym zwykle jest wykorzystywana, z uwzględnieniem toksycznego wpływu na ludzi, zwierzęta i inne organizmy (w odniesieniu do aspektów bezpieczeństwa dla środowiska)
3. CHARAKTERYSTYKA MOLEKULARNA
3.1. Informacje dotyczące modyfikacji genetycznej
a) opis metod zastosowanych w celu dokonania modyfikacji genetycznej
b) rodzaj i źródło wykorzystywanego wektora
c) źródło kwasu(-ów) nukleinowego(-ych) dawcy wykorzystywanego(-ych) dla celów transformacji, wielkość i zamierzona funkcja każdego fragmentu stanowiącego część składową regionu przeznaczonego do insercji
3.2. Informacje o roślinie zmodyfikowanej genetycznie
3.2.1. Opis cech i właściwości, które zostały wprowadzone lub zmienione
3.2.2. Informacje dotyczące sekwencji kwasów nukleinowych, które zostały faktycznie dodane lub usunięte
a) liczba kopii wszystkich wykrywalnych insertów, zarówno kompletnych, jak i częściowych
b) w przypadku delecji – wielkość i funkcja usuniętego regionu (regionów)
c) subkomórkowe lokalizacje wprowadzonego insertu (jądro, chloroplasty, mitochondria lub pozostające w formie niezintegrowanej) oraz metody ich określenia
d) organizacja wprowadzonego materiału genetycznego w miejscu inercji
e) w przypadku modyfikacji innych niż insercja lub delecja opisać funkcję zmodyfikowanego materiału genetycznego przed modyfikacją i po niej, jak również bezpośrednie zmiany w ekspresji genów w wyniku modyfikacji
3.2.3. Informacje na temat ekspresji insertu
a) informacje dotyczące rozwojowej ekspresji wprowadzonego insertu podczas cyklu życiowego rośliny
b) części rośliny, w których dochodzi do ekspresji insertu
3.2.4. Stabilność genetyczna insertu i stabilność fenotypowa rośliny zmodyfikowanej genetycznie
3.2.5. Informacje dotyczące różnic między rośliną zmodyfikowaną genetycznie a rośliną biorcą (w odniesieniu do aspektów bezpieczeństwa dla środowiska) pod względem:
a) sposobów lub skali rozmnażania
b) rozprzestrzeniania
c) zdolności przetrwania
d) innych różnic
3.2.6. Wszelkie zmiany, jakie zaszły w roślinie zmodyfikowanej genetycznie w zakresie zdolności do transferu materiału genetycznego do innych organizmów (w odniesieniu do aspektów bezpieczeństwa dla środowiska)
a) transfer genów z rośliny do bakterii
b) transfer genów z rośliny do rośliny
4. ANALIZA PORÓWNAWCZA
4.1. Wybór tradycyjnego odpowiednika i dodatkowych okazów porównawczych
4.2. Schemat doświadczenia i statystyczna analiza danych z doświadczeń polowych na potrzeby analizy porównawczej
Opis schematu doświadczenia (liczba lokalizacji, sezony wegetacyjne, rozprzestrzenienie geograficzne, replikacja i liczba odmian handlowych w każdej lokalizacji) i analizy statystycznej.
4.3. Wybór materiału i związków do analizy
4.4. Analiza porównawcza właściwości agronomicznych i fenotypowych
4.5. Skutki przetwarzania
5. TOKSYKOLOGIA
a) toksykologiczne badanie nowych białek ulegających ekspresji
b) badanie nowych składników innych niż białka
c) informacje na temat naturalnych składników żywności lub paszy
d) badanie całej genetycznie zmodyfikowanej żywności i paszy
6. ALERGENNOŚĆ
a) ocena alergenności nowych białek ulegających ekspresji
b) ocena alergenności całej rośliny zmodyfikowanej genetycznie
7. OCENA WARTOŚCI ODŻYWCZEJ
a) ocena wartości odżywczych genetycznie zmodyfikowanej żywności
b) ocena wartości odżywczych genetycznie zmodyfikowanej paszy
8. OCENA NARAŻENIA – PRZEWIDYWANE SPOŻYCIE/ZAKRES ZASTOSOWANIA
9. CHARAKTERYSTYKA RYZYKA
10. MONITOROWANIE GENETYCZNIE ZMODYFIKOWANEJ ŻYWNOŚCI LUB PASZY PO WPROWADZENIU DO OBROTU
11. OCENA ODDZIAŁYWANIA NA ŚRODOWISKO
11.1. Mechanizm wzajemnego oddziaływania między rośliną zmodyfikowaną genetycznie a organizmami docelowymi
11.2. Potencjalne zmiany wzajemnych oddziaływań między rośliną zmodyfikowaną genetycznie a środowiskiem biotycznym, wynikające z modyfikacji genetycznej
a) trwałość i inwazyjność
b) wybiórcze korzyści lub szkody
c) zdolność transferu genów
d) wzajemne oddziaływanie między rośliną zmodyfikowaną genetycznie a organizmami docelowymi
e) wzajemne oddziaływanie między rośliną zmodyfikowaną genetycznie a organizmami niedocelowymi
f) wpływ na zdrowie człowieka
g) wpływ na zdrowie zwierząt
h) skutki dla procesów biogeochemicznych
i) wpływ poszczególnych technik uprawy, zarządzania i zbioru
11.3. Potencjalne wzajemne oddziaływania ze środowiskiem abiotycznym
11.4. Charakterystyka ryzyka
12. PLAN MONITOROWANIA SKUTKÓW DLA ŚRODOWISKA
a) informacje ogólne (ocena ryzyka, podstawowe informacje)
b) wzajemna zależność między oceną ryzyka dla środowiska naturalnego a monitorowaniem
c) monitorowanie konkretnych przypadków rośliny zmodyfikowanej genetycznie (podejście, strategia, metoda i analiza)
d) ogólny nadzór nad wpływem rośliny zmodyfikowanej genetycznie (podejście, strategia, metoda i analiza)
e) składanie sprawozdań z wyników monitorowania
13. TECHNIKI WYKRYWANIA I IDENTYFIKACJI ROŚLINY ZMODYFIKOWANEJ GENETYCZNIE
14. INFORMACJE DOTYCZĄCE WCZEŚNIEJSZYCH UWOLNIEŃ ROŚLINY ZMODYFIKOWANEJ GENETYCZNIE (W ODNIESIENIU DO ASPEKTÓW BEZPIECZEŃSTWA DLA ŚRODOWISKA)
14.1. Historia wcześniejszych uwolnień rośliny zmodyfikowanej genetycznie zgłoszonej zgodnie z częścią B dyrektywy 2001/18/WE lub częścią B dyrektywy Rady 90/220/EWG (3) przez tego samego zgłaszającego
a) numer zgłoszenia
b) wnioski z monitorowania po uwolnieniu
c) wyniki uwolnienia w odniesieniu do jakiegokolwiek ryzyka dla zdrowia ludzi i dla środowiska, przedłożone właściwemu organowi zgodnie z art. 10 dyrektywy 2001/18/WE
14.2. Historia wcześniejszych uwolnień rośliny zmodyfikowanej genetycznie przeprowadzonych poza terytorium Unii przez tego samego zgłaszającego
a) kraj, w którym dokonano uwolnienia
b) organ nadzorujący uwolnienie
c) miejsce uwolnienia
d) cel uwolnienia
e) czas trwania uwolnienia
f) cel monitorowania po uwolnieniu
g) czas trwania monitorowania po uwolnieniu
h) wnioski z monitorowania po uwolnieniu
i) wyniki uwolnienia w odniesieniu do jakiegokolwiek ryzyka dla zdrowia ludzi i dla środowiska
(1) Dziennik EFSA 2010; 8(6):1637.
(2) Dz.U. L 287 z 5.11.2003, s. 1.
(3) Dz.U. L 117 z 8.5.1990, s. 15.
ZAŁĄCZNIK II
WYMOGI NAUKOWE DOTYCZĄCE OCENY RYZYKA GENETYCZNIE ZMODYFIKOWANEJ ŻYWNOŚCI I PASZY
I. WPROWADZENIE
1. DEFINICJE
Do celów niniejszego załącznika stosuje się następujące definicje:
1) „identyfikacja zagrożeń” oznacza identyfikację czynników biologicznych, chemicznych i fizycznych, które mogą mieć negatywne skutki dla zdrowia i które mogą występować w określonej żywności i paszy lub w grupie żywności i pasz;
2) „charakterystyka zagrożeń” oznacza jakościową lub ilościową ocenę charakteru szkodliwych skutków dla zdrowia związanych z czynnikami biologicznymi, chemicznymi i fizycznymi, które mogą być obecne w żywności i paszy;
3) „ charakterystyka ryzyka” oznacza oszacowanie jakościowe lub ilościowe, w tym określenie towarzyszących niepewności, prawdopodobieństwa wystąpienia i dotkliwości znanych lub potencjalnych szkodliwych skutków dla zdrowia w danej populacji w oparciu o identyfikację zagrożeń, charakterystykę zagrożeń i ocenę narażenia.
2. UWAGI SZCZEGÓŁOWE
2.1. Insercja genów markerowych i innych sekwencji kwasu(-ów) nukleinowego(-ych), która nie jest niezbędna do uzyskania pożądanej cechy
Aby ułatwić ocenę ryzyka, wnioskodawca podejmuje starania w celu zminimalizowania obecności wprowadzonych sekwencji kwasu(-ów) nukleinowego(-ych), które nie są niezbędne do uzyskania pożądanej cechy.
Podczas procesu modyfikacji genetycznej roślin i innych organizmów geny markerowe są często wykorzystywane do ułatwienia wyboru i identyfikacji genetycznie zmodyfikowanych komórek, zawierających przedmiotowy gen wprowadzony do genomu organizmu gospodarza, wśród ogromnej większości komórek nieprzekształconych. Wnioskodawca starannie wybiera takie geny markerowe, przestrzegając przepisów art. 4 ust. 2 dyrektywy 2001/18/WE. W tym kontekście celem wnioskodawcy jest opracowanie organizmów zmodyfikowanych genetycznie bez korzystania z genów markerowych kodujących oporność na antybiotyki.
2.2. Ocena ryzyka stwarzanego przez genetycznie zmodyfikowaną żywność i paszę zawierające złożone modyfikacje genetyczne
Na potrzeby oceny ryzyka stwarzanego przez genetycznie zmodyfikowaną żywność i paszę zawierające złożone modyfikacje genetyczne, uzyskane poprzez konwencjonalne krzyżowanie roślin zmodyfikowanych genetycznie zawierających jedną lub kilka modyfikacji genetycznych, wnioskodawca przedkłada ocenę ryzyka dotyczącą każdej pojedynczej modyfikacji genetycznej lub, zgodnie z art. 3 ust. 6 niniejszego rozporządzenia, czyni odniesienie do wcześniej złożonego(-ych) wniosku(-ów). Ocena ryzyka stwarzanego przez genetycznie zmodyfikowaną żywność i paszę zawierające złożone modyfikacje genetyczne obejmuje również ocenę następujących aspektów:
a) stabilności modyfikacji genetycznych;
b) ekspresji modyfikacji genetycznych;
c) potencjalne efekty synergiczne lub antagonistyczne wynikające z kombinacji modyfikacji genetycznych podlegają ocenie zgodnie z sekcjami 1.4 (Toksykologia), 1.5 (Alergenność) i 1.6 (Ocena wartości odżywczych).
W przypadku genetycznie zmodyfikowanej żywności i paszy zawierających rośliny zmodyfikowane genetycznie, składających się z nich lub produkowanych z takich roślin, których uprawa związana jest z produkcją materiału genetycznie zmodyfikowanego zawierającego różne subkombinacje modyfikacji genetycznych (segregacja upraw), wniosek obejmuje wszystkie subkombinacje niezależnie od ich pochodzenia, które nie zostały jeszcze zatwierdzone. W takim przypadku wnioskodawca przedstawia powody naukowe uzasadniające brak potrzeby przedstawienia danych doświadczalnych dla przedmiotowych subkombinacji lub, w przypadku braku takich powodów, przedstawia dane doświadczalne.
W przypadku zmodyfikowanej żywności i paszy zawierających rośliny zmodyfikowane genetycznie, składających się z nich lub produkowanych z takich roślin, których uprawa nie prowadzi do produkcji materiału genetycznie zmodyfikowanego zawierającego różne kombinacje modyfikacji genetycznych (brak segregacji upraw), wniosek obejmuje tylko tę kombinację, która ma zostać wprowadzona do obrotu.
Zasady określone w niniejszej sekcji stosuje się odpowiednio do modyfikacji genetycznych, które są połączone za pomocą innych środków, takich jak kotransformacja i retransformacja.
II. WYMOGI NAUKOWE
1. IDENTYFIKACJA I CHARAKTERYSTYKA ZAGROŻEŃ
1.1. Informacje dotyczące roślin biorców lub (w stosownych przypadkach) roślin rodzicielskich
1.1.1. Wnioskodawca przedstawia wyczerpujące informacje dotyczące roślin biorców lub (w stosownych przypadkach) roślin rodzicielskich w celu:
a) dokonania oceny wszystkich kwestii, które mogą budzić obawy, takich jak obecność naturalnych toksyn lub alergenów;
b) określenia potrzeby przeprowadzenia konkretnych analiz.
1.1.2. Dla celów, o których mowa w pkt 1.1.1, wnioskodawca podaje następujące informacje:
a) pełna nazwa:
(i) rodzina;
(ii) rodzaj;
(iii) gatunek;
(iv) podgatunek;
(v) kultywar/linia hodowlana lub szczep;
(vi) nazwa zwyczajowa;
b) geograficzne rozmieszczenie i uprawa rośliny, w tym rozmieszczenie w obrębie Unii;
c) informacje na temat roślin biorców lub roślin rodzicielskich istotne dla ich bezpieczeństwa, w tym na temat znanej toksyczności lub alergenności;
d) dane dotyczące wcześniejszego i obecnego zastosowania rośliny biorcy. Informacje te obejmują historię bezpiecznego stosowania do spożycia jako żywność lub pasza, informacje na temat zwykłych sposobów uprawiania, transportowania i przechowywania danej rośliny, tego, czy potrzebne jest specjalne przetwarzanie, aby można było bezpiecznie spożywać daną roślinę, opis normalnej roli danej rośliny w diecie (np. która część rośliny jest wykorzystywana jako źródło żywności lub paszy, czy jej spożycie jest ważne w szczególnych podgrupach populacji, jakie ważne makro- lub mikroskładniki pokarmowe wnosi do diety).
1.2. Charakterystyka molekularna
1.2.1. Informacje dotyczące modyfikacji genetycznej
Wnioskodawca przedstawia wystarczające informacje na temat modyfikacji genetycznej:
a) w celu określenia kwasu(-ów) nukleinowego(-ych) przeznaczonego(-ych) do transformacji i powiązanych sekwencji wektorów potencjalnie wprowadzonych do rośliny biorcy;
b) w celu scharakteryzowania kwasu(-ów) nukleinowego(-ych) faktycznie wprowadzonego(-ych) do rośliny.
1.2.1.1. Opis metod zastosowanych w celu dokonania modyfikacji genetycznej
Wnioskodawca przedstawia informacje na temat poniższych elementów:
a) metody transformacji genetycznej, w tym odpowiednie odniesienia;
b) materiał rośliny biorcy;
c) gatunek i szczep Agrobacterium i innych mikroorganizmów, jeżeli są wykorzystywane w procesie transformacji genetycznej;
d) plazmidy pomocnicze, jeżeli są wykorzystywane w procesie transformacji genetycznej;
e) źródła nośnikowego(-ych) kwasu(-ów) nukleinowego(-ych), jeżeli jest (są) wykorzystywany(-e) w procesie transformacji genetycznej.
1.2.1.2. Rodzaj i źródło wykorzystywanego wektora
Wnioskodawca przedstawia następujące informacje:
a) fizyczna mapa elementów funkcjonalnych i innych składników plazmidów/wektorów wraz z istotnymi informacjami niezbędnymi do interpretacji analiz molekularnych (np. miejsca restrykcji, pozycje primerów wykorzystywanych w łańcuchowej reakcji polimerazy (PCR), położenie sond stosowanych w analizie typu Southern). Wyraźnie wskazuje się region przeznaczony do insercji;
b) tabela z wyszczególnieniem każdego składnika plazmidu/wektora (w tym region przeznaczony do insercji), jego rozmiar, pochodzenie i zamierzona funkcja.
1.2.1.3. Źródło kwasu(-ów) nukleinowego(-ych) wykorzystanego(-ych) dla celów transformacji, wielkość i zamierzona funkcja każdego fragmentu stanowiącego część składową regionu przeznaczonego do insercji
W celu określenia, czy charakter organizmu dawcy lub sekwencji kwasu(-ów) nukleinowego(-ych) mogą wywołać problemy związane z bezpieczeństwem, wnioskodawca przedstawia informacje na temat organizmu dawcy i sekwencji kwasu(-ów) nukleinowego(-ych) przeznaczonej do insercji.
Informacje dotyczące funkcji regionów kwasu nukleinowego przeznaczonych do insercji obejmują następujące elementy:
a) pełna sekwencja kwasu(-ów) nukleinowego(-ych) przeznaczona do insercji, w tym informacje na temat wszelkich zamierzonych zmian w odpowiednich sekwencjach w organizmach dawców;
b) historia bezpiecznego stosowania produktów genowych powstałych z regionów przeznaczonych do insercji;
c) dane na temat możliwego związku produktów genowych ze znanymi toksynami, substancjami obniżającymi właściwości odżywcze pokarmów i alergenami.
Informacje dotyczące każdego organizmu dawcy obejmują:
– klasyfikację taksonomiczną,
– historię stosowania w odniesieniu do bezpieczeństwa żywności i paszy.
1.2.2. Informacje o roślinie zmodyfikowanej genetycznie
1.2.2.1. Ogólny opis cech i właściwości, które zostały wprowadzone lub zmienione
Informacje podane w niniejszym punkcie mogą być ograniczone do ogólnego opisu wprowadzonych cech i wynikających z nich zmian fenotypu i metabolizmu rośliny.
Na przykład jeżeli wprowadzoną cechą jest tolerancja na herbicydy, wnioskodawca przedstawia informacje na temat sposobu działania substancji czynnej oraz metabolizmu tej substancji w roślinie.
1.2.2.2. Informacje dotyczące sekwencji, które zostały faktycznie dodane/usunięte
Wnioskodawca przedstawia następujące informacje:
a) rozmiar i liczbę kopii wszystkich wykrywalnych insertów, zarówno kompletnych, jak i częściowych; ustala się to zwykle poprzez analizę typu Southern.
Kombinacje sondy/enzymu restrykcyjnego stosowane w tym celu zapewniają całkowite uwzględnienie sekwencji, które mogły zostać wprowadzone do rośliny zmodyfikowanej genetycznie, takich jak jakiekolwiek części plazmidu/wektora lub jakiekolwiek nośnikowe lub obce kwasy nukleinowe pozostające w roślinie zmodyfikowanej genetycznie.
Analiza typu Southern obejmuje całe transgeniczne locus (loci), jak również sekwencje flankujące i uwzględnia wszystkie odpowiednie kontrole.
W celu określenia liczby kopii insertu można wykorzystać również metody uzupełniające (takie jak PCR w czasie rzeczywistym);
b) organizację i sekwencję wprowadzonego materiału genetycznego w każdym miejscu insercji, w standardowym formacie elektronicznym, w celu identyfikacji zmian we wprowadzonych sekwencjach w porównaniu z sekwencją przeznaczoną do insercji;
c) w przypadku delecji – wielkość i funkcję usuniętego regionu (regionów), jeśli jest to możliwe;
d) subkomórkowe lokalizacje insertów (jądro, chloroplasty, mitochondria lub pozostające w formie niezintegrowanej) i metody ich określenia;
e) informacje na temat sekwencji regionów flankujących 5’ i 3’ każdego miejsca insercji, w standardowym formacie elektronicznym, w celu zidentyfikowania przerwań znanych genów.
Analizy bioinformatyczne przeprowadza się przy zastosowaniu aktualnych baz danych w celu wyszukania podobieństw zarówno wewnątrzgatunkowych, jak i międzygatunkowych.
W przypadku zmodyfikowanych genetycznie roślin zawierających złożone modyfikacje genetyczne ocenia się bezpieczeństwo ewentualnych interakcji między wszelkimi niezamierzonymi modyfikacjami w każdym miejscu insercji;
f) otwarte ramki odczytu (zwane dalej „ORF” i definiowane jako każda sekwencja nukleotydu zawierająca ciąg kodonów, który nie jest przerwany przez obecność kodonu STOP w tej samej ramce odczytu) utworzone w wyniku modyfikacji genetycznej albo w miejscach łączenia z DNA genomowym, albo z powodu wewnętrznych rearanżacji insertów.
ORF analizuje się pomiędzy kodonami STOP, bez ograniczania ich długości. Analizy bioinformatyczne przeprowadza się przy zastosowaniu aktualnych baz danych w celu zbadania możliwych podobieństw ze znanymi toksynami lub alergenami.
Podaje się cechy i wersje baz danych.
W zależności od zgromadzonych informacji ukończenie oceny zagrożeń może wymagać przeprowadzenia dalszych analiz (np. analizy transkrypcji).
1.2.2.3. Informacje dotyczące ekspresji insertu
Wnioskodawca przedstawia informacje:
– w celu wykazania, czy wprowadzona/zmodyfikowana sekwencja wywołuje zamierzone zmiany na poziomie białka, RNA lub metabolitu,
– w celu scharakteryzowania potencjalnej niezamierzonej ekspresji nowych ORF, co do których w ramach pkt 1.2.2.2 lit. f) stwierdzono, że budzą obawy w kwestii bezpieczeństwa.
W tym celu wnioskodawca przedstawia następujące informacje:
a) metody zastosowane na potrzeby analizy ekspresji wraz z charakterystyką ich wydajności;
b) informacje dotyczące rozwojowej ekspresji insertu podczas cyklu życiowego rośliny.
Wymóg dotyczący informacji dotyczących rozwojowej ekspresji rozpatruje się oddzielnie dla każdego przypadku biorąc pod uwagę zastosowany promotor, zamierzony skutek modyfikacji i zakres wniosku;
c) części rośliny, w których dochodzi do ekspresji insertu/zmodyfikowanych sekwencji;
d) potencjalną niezamierzoną ekspresję nowych ORF zidentyfikowaną w ramach pkt 1.2.2.2 lit. f), która budzi obawy w kwestii bezpieczeństwa;
e) dane dotyczące ekspresji białek, w tym dane nieprzetworzone, uzyskane z doświadczeń polowych i związane z warunkami, w których roślina jest uprawiana.
We wszystkich przypadkach podaje się dane dotyczące poziomów ekspresji z tych części rośliny, które wykorzystywane są w żywności i paszy.
Ponadto podaje się również informacje na temat ekspresji genów docelowych w innych częściach rośliny, jeżeli zastosowano promotory tkankowo swoiste i gdy jest to istotne dla oceny bezpieczeństwa. Minimalnym wymogiem w odniesieniu do ekspresji białka jest podanie danych pochodzących z trzech miejsc uprawy lub z jednego miejsca, ale zebranych w ciągu trzech sezonów. Kombinacje miejsc i sezonów są dopuszczalne, pod warunkiem że spełniony jest minimalny wymóg. W przypadkach uzasadnionych charakterem insertu (takich jak podejście wyciszające lub gdy szlaki biochemiczne zostały celowo zmienione) analizuje się określone RNA lub metabolity.
W przypadku podejścia wyciszającego poprzez ekspresję RNAi ewentualnych genów niedocelowych należy szukać w drodze analizy in silico, aby ocenić, czy modyfikacja genetyczna może wpływać na ekspresję innych genów, która budzi obawy w kwestii bezpieczeństwa;
f) w odniesieniu do łączenia modyfikacji genetycznych poprzez konwencjonalne krzyżowanie podaje się dane dotyczące ekspresji w celu oceny ewentualnych interakcji między modyfikacjami, które mogą budzić dodatkowe obawy w kwestii bezpieczeństwa w stosunku do ekspresji białka i cechy w porównaniu z pojedynczymi modyfikacjami genetycznymi. Porównanie przeprowadza się na podstawie danych uzyskanych z roślin uprawianych w ramach tych samych doświadczeń polowych. W poszczególnych przypadkach i gdy istnieją obawy, konieczne mogą być dodatkowe informacje.
1.2.2.4. Stabilność genetyczna insertu i stabilność fenotypowa rośliny zmodyfikowanej genetycznie
Wnioskodawca przedstawia informacje:
a) w celu wykazania stabilności genetycznej transgenicznych loci oraz stabilności fenotypowej i wzoru dziedziczenia wprowadzonych cech;
b) w przypadku złożonych modyfikacji genetycznych – w celu ustalenia, że każda ze złożonych modyfikacji genetycznych w roślinie posiada takie same właściwości i cechy molekularne, jak w roślinach zawierających pojedyncze modyfikacje genetyczne.
Na potrzeby tych informacji wnioskodawca przekazuje dane, które wykazują stabilność w wielu (zazwyczaj pięciu) pokoleniach lub cyklach wegetacyjnych w odniesieniu do roślin zawierających pojedyncze modyfikacje genetyczne. Wystarczające są dane z pierwszego i ostatniego pokolenia cyklów wegetacyjnych. Podaje się źródło materiału używanego w analizie. Dane są analizowane przy użyciu właściwych metod statystycznych.
W przypadku złożonych modyfikacji genetycznych porównania między oryginalnymi modyfikacjami genetycznymi a złożonymi modyfikacjami genetycznymi przeprowadza się z wykorzystaniem materiałów roślinnych reprezentatywnych w stosunku do roślin przeznaczonych do produkcji komercyjnej. Wnioskodawca przedstawia odpowiednie uzasadnienie dotyczące użytych materiałów roślinnych. Porównania obejmują porównania sekwencji insertów i regionów flankujących uzyskanych z roślin zmodyfikowanych genetycznie zawierających pojedyncze modyfikacje i z roślin zawierających złożone modyfikacje genetyczne.
Aby ocenić stabilność genetyczną modyfikacji genetycznych, wnioskodawca stosuje odpowiednie rodzaje podejścia molekularnego, o których mowa w sekcji 1.2.2.2.
1.2.2.5. Potencjalne ryzyko związane z poziomym transferem genów
Wnioskodawca ocenia prawdopodobieństwo poziomego transferu genów z produktu na ludzi, zwierzęta i mikroorganizmy oraz wszelkie ewentualne powiązane ryzyko, gdy nienaruszone i funkcjonalne kwasy nukleinowe pozostają w genetycznie zmodyfikowanej żywności i paszy.
1.2.3. Wnioski z charakterystyki molekularnej
Charakterystyka molekularna dostarcza danych na temat struktury i ekspresji insertów oraz stabilności zamierzonych cech. Dotyczy to również sytuacji, w których modyfikacje genetyczne zostały połączone poprzez tradycyjne metody rozmnażania.
W szczególności podaje się, czy charakterystyka molekularna modyfikacji genetycznych wywołuje obawy związane z bezpieczeństwem w odniesieniu do przerwania genów endogenicznych lub sekwencji regulacyjnych.
Charakterystyka molekularna ma również na celu określenie, czy modyfikacje genetyczne budzą obawy w odniesieniu do zdolności produkowania białek/substancji innych niż zamierzone, a szczególnie nowych toksyn lub alergenów.
Potencjalne niezamierzone zmiany określone w niniejszej sekcji uwzględnia się w odpowiednich uzupełniających częściach oceny bezpieczeństwa.
1.3. Analiza porównawcza
Analiza porównawcza składu oraz właściwości agronomicznych i fenotypowych stanowi wraz z charakterystyką molekularną punkt wyjścia dla zorganizowania i przeprowadzenia oceny ryzyka dotyczącej nowej genetycznie zmodyfikowanej żywności i paszy.
Jest ona ukierunkowana na określenie podobieństw i różnic:
a) w składzie oraz właściwościach agronomicznych i fenotypowych (zmiany zamierzone i niezamierzone) między rośliną zmodyfikowaną genetycznie a ich tradycyjnym odpowiednikiem;
b) w składzie między genetycznie zmodyfikowaną żywnością i paszą a ich tradycyjnym odpowiednikiem.
Jeżeli nie można określić odpowiedniego tradycyjnego odpowiednika, nie można przeprowadzić porównawczej oceny bezpieczeństwa, a zatem ocenę bezpieczeństwa i wartości odżywczych genetycznie zmodyfikowanej żywności lub paszy przeprowadza się jak w przypadku nowej żywności objętej zakresem stosowania rozporządzenia (WE) nr 258/97 Parlamentu Europejskiego i Rady (1), która to żywność nie ma tradycyjnych odpowiedników (podobnie jak w przypadku, gdy genetycznie zmodyfikowana żywność lub pasza nie są ściśle związane z żywnością lub paszą posiadającymi historię bezpiecznego stosowania lub gdy wprowadza się określoną cechę lub określone cechy z zamiarem doprowadzenia do złożonych zmian w składzie genetycznie zmodyfikowanej żywności lub paszy).
1.3.1. Wybór tradycyjnego odpowiednika i dodatkowych okazów porównawczych
W przypadku upraw rozmnażanych wegetatywnie tradycyjnym odpowiednikiem jest z zasady odmiana blisko izogeniczna wykorzystywana do tworzenia linii transgenicznej.
W przypadku upraw rozmnażanych płciowo tradycyjny odpowiednik ma podłoże genetyczne porównywalne z rośliną zmodyfikowaną genetycznie. W przypadku gdy roślina zmodyfikowana genetycznie została rozmnożona za pomocą krzyżowania wstecznego, wybiera się tradycyjny odpowiednik o podłożu genetycznym jak najbardziej zbliżonym do rośliny zmodyfikowanej genetycznie.
Ponadto wnioskodawca może uwzględnić okaz porównawczy o podłożu genetycznym bardziej zbliżonym do rośliny zmodyfikowanej genetycznie niż w przypadku tradycyjnego odpowiednika (np. negatywny segregant).
W przypadku roślin zmodyfikowanych genetycznie tolerujących herbicydy oraz w celu dokonania oceny, czy oczekiwane sposoby prowadzenia działalności rolniczej wpływają na ekspresję badanych punktów końcowych, porównuje się trzy materiały badawcze: roślinę zmodyfikowaną genetycznie wystawioną na działanie zamierzonego herbicydu, tradycyjny odpowiednik objęty konwencjonalnymi systemami traktowania herbicydami oraz roślinę zmodyfikowaną genetycznie objętą tymi samymi konwencjonalnymi systemami traktowania herbicydami.
W przypadku złożonych modyfikacji genetycznych nie zawsze można zastosować tradycyjny odpowiednik, który byłby tak zbliżony pod względem genetycznym do rośliny zmodyfikowanej genetycznie jak tradycyjny odpowiednik zwykle stosowany w przypadku pojedynczych modyfikacji genetycznych. W takich okolicznościach wnioskodawca przedstawia uzasadnienie wyboru tradycyjnego odpowiednika i ocenia jego ograniczenia w zakresie oceny ryzyka. Ponadto jako dodatkowe okazy porównawcze można również uwzględnić pojedyncze genetycznie zmodyfikowane linie rodzicielskie lub genetycznie zmodyfikowane linie zawierające subkombinację złożonych modyfikacji genetycznych, w odniesieniu do których złożono wniosek, lub negatywne segreganty pochodzące z tych genetycznie zmodyfikowanych linii. Wnioskodawca podaje szczegółowe informacje uzasadniające wybór dodatkowych okazów porównawczych.
We wszystkich przypadkach wnioskodawca podaje informacje na temat programu rozmnażania (rodowodu) w odniesieniu do rośliny zmodyfikowanej genetycznie, tradycyjnego odpowiednika i, w stosownych przypadkach, dodatkowych okazów porównawczych, wraz z odpowiednim uzasadnieniem ich wyboru. Historia bezpiecznego stosowania tradycyjnego odpowiednika jest odpowiednio poparta danymi zarówno jakościowymi, jak i ilościowymi.
Bardziej szczegółowe wytyczne dotyczące stosowania wymogów zawartych w niniejszej sekcji są dostępne w opinii naukowej EFSA pt. „Guidance on selection of comparators for the risk assessment of genetically modified plants and derived food and feed” (Wytyczne dotyczące wyboru okazów porównawczych na potrzeby oceny ryzyka w odniesieniu do roślin zmodyfikowanych genetycznie oraz pochodnej żywności i paszy) (2).
1.3.2. Schemat doświadczenia i statystyczna analiza danych z doświadczeń polowych na potrzeby analizy porównawczej
1.3.2.1. Opis protokołów schematu doświadczenia
a) Zasady schematu doświadczenia
Doświadczenia polowe wykorzystywane do produkcji materiału na potrzeby analizy porównawczej są wykonywane w celu ustalenia, czy roślina zmodyfikowana genetycznie lub zmodyfikowana genetycznie żywność i pasza różnią się od swoich tradycyjnych odpowiedników lub są równoważne z niezmodyfikowanymi genetycznie odmianami wzorcowymi o historii bezpiecznego stosowania.
Dla każdego punktu końcowego analiza porównawcza obejmuje następujące dwa rodzaje podejścia:
(i) badanie różnicy, aby sprawdzić, czy roślina zmodyfikowana genetycznie różni się od swojego tradycyjnego odpowiednika i może w związku z tym zostać uznana za zagrożenie w zależności od rodzaju stwierdzonej różnicy, a także od wielkości i typu narażenia;
(ii) badanie równoważności, aby sprawdzić, czy oprócz wprowadzonej cechy (cech) roślina zmodyfikowana genetycznie jest równoważna niezmodyfikowanym genetycznie odmianom wzorcowym, czy nie.
W badaniu różnicy zakłada się w hipotezie zerowej, że nie ma różnicy między GMO a jego tradycyjnym odpowiednikiem, natomiast w hipotezie alternatywnej zakłada się, że różnica istnieje.
W przypadku gdy do oceny ryzyka wykorzystuje się dodatkowe okazy porównawcze, przeprowadza się badanie różnicy między rośliną zmodyfikowaną genetycznie a każdym z dodatkowych okazów porównawczych, zgodnie z wymogami przedstawionymi w sekcji 1.3.2.2 dla badania różnicy między rośliną zmodyfikowaną genetycznie a jej tradycyjnym odpowiednikiem.
W badaniu równoważności zakłada się w hipotezie zerowej, że różnica między GMO a zbiorem odmian wzorcowych jest co najmniej tak duża, jak określona wielkość minimalna (zob. sekcja 1.3.2.2), natomiast w hipotezie alternatywnej zakłada się, że między GMO a zbiorem odmian wzorcowych nie ma różnicy lub że istnieje różnica mniejsza niż określona wielkość minimalna.
Do stwierdzenia, że GMO i zbiór odmian wzorcowych są jednoznacznie równoważne w odniesieniu do badanego punktu końcowego, wymagane jest odrzucenie hipotezy zerowej. Granice równoważności stosowane w badaniu równoważności oznaczają odpowiednio zakres naturalnej zmienności oczekiwanej w przypadku odmian wzorcowych posiadających historię bezpiecznego stosowania.
b) Szczegółowe protokoły schematu doświadczenia
Naturalna zmienność może mieć kilka źródeł: zmienność w obrębie odmiany powstaje w wyniku czynników środowiskowych, a zmienność między odmianami powstaje w wyniku połączenia zarówno czynników genetycznych, jak i środowiskowych. W celu określenia i oszacowania różnic związanych jedynie z genotypami istotne jest kontrolowanie zmienności środowiskowej. Z tego względu niezmodyfikowane genetycznie odmiany wzorcowe włącza się do schematu doświadczenia dotyczącego doświadczeń polowych i uwzględnia się wystarczającą liczbę tych odmian, aby zapewnić odpowiednie oszacowanie zmienności wymaganej do określenia granic równoważności. Wszystkie materiały badawcze składające się z roślin zmodyfikowanych genetycznie, tradycyjnego odpowiednika, odmian wzorcowych i, w stosownych przypadkach, dodatkowych okazów porównawczych, są losowo przydzielane do działek w obrębie jednego pola w każdym miejscu, zwykle w całkowicie zrandomizowanym lub zrandomizowanym blokowym schemacie doświadczenia. Różne miejsca wybrane do doświadczeń polowych odzwierciedlają różne warunki meteorologiczne i agronomiczne, w których rośliny mają być uprawiane; wybór ten jest wyraźnie uzasadniany. Wybór niezmodyfikowanych genetycznie odmian wzorcowych jest odpowiedni do wybranych miejsc i wyraźnie uzasadniany. W przypadku gdy miejsca obejmują ograniczony zakres warunków uprawy, wnioskodawca powtarza doświadczenia polowe przez okres dłuższy niż jeden rok.
W obrębie każdego miejsca materiały badawcze składające się z roślin zmodyfikowanych genetycznie, tradycyjnego odpowiednika i, w stosownych przypadkach, dodatkowych okazów porównawczych są identyczne dla wszystkich replikacji. Ponadto w każdym miejscu znajdują się co najmniej trzy odpowiednie niezmodyfikowane genetycznie odmiany wzorcowe danej rośliny, które posiadają znaną historię bezpiecznego użytkowania i które również są identyczne w poszczególnych replikacjach, chyba że istnieje wyraźne uzasadnienie dla niespełnienia tego warunku. Replikacja w każdym miejscu to liczba wyników uzyskanych dla każdego materiału badawczego; replikacja nie powinna nigdy wynosić mniej niż cztery w każdym z miejsc. Jeśli jednak w danym miejscu dostępne są jedynie dwie odpowiednie odmiany wzorcowe, replikacja w tym miejscu musi wynosić sześć; jeśli dostępna jest tylko jedna odmiana wzorcowa, replikacja wynosi osiem.
Każde doświadczenie polowe jest replikowane w co najmniej ośmiu miejscach, wybranych jako reprezentatywne dla różnych możliwych środowisk przyjmujących, w których dana roślina ma być uprawiana. Doświadczenia polowe mogą być przeprowadzane w jednym roku lub rozłożone na wiele lat. Odmiany wzorcowe niezmodyfikowane genetycznie mogą różnić się w zależności od miejsca. W całym zbiorze doświadczeń polowych używa się co najmniej sześciu różnych odmian wzorcowych.
W przypadku gdy roślina zmodyfikowana genetycznie jest badana wraz z innymi roślinami zmodyfikowanymi genetycznie tego samego gatunku (np. Zea mays), materiał na potrzeby oceny porównawczej tych różnych roślin zmodyfikowanych genetycznie może być produkowany równolegle w tym samym miejscu i w ramach tego samego doświadczenia polowego, przez umieszczenie różnych roślin zmodyfikowanych genetycznie i ich odpowiednich okazów porównawczych w tym samym bloku zrandomizowanym. Podlega to następującym dwóm rygorystycznym warunkom:
(i) tradycyjny odpowiednik oraz, w stosownych przypadkach, dodatkowe okazy porównawcze zawsze występują w tym samym bloku razem z rośliną zmodyfikowaną genetycznie;
(ii) wszystkie różne rośliny zmodyfikowane genetycznie i ich okazy porównawcze oraz wszystkie niezmodyfikowane genetycznie odmiany wzorcowe wykorzystywane do badania równoważności z tymi roślinami zmodyfikowanymi genetycznie są całkowicie zrandomizowane w każdym bloku.
W przypadku gdy liczba działek w bloku potrzebna do przeprowadzenia takiego doświadczenia polowego przekracza 16, można zastosować układ bloków niekompletnych częściowo zrównoważonych, aby zmniejszyć liczbę działek w bloku przez wyłączenie z każdego bloku niektórych z roślin zmodyfikowanych genetycznie i ich odpowiednich okazów porównawczych. Podlega to następującym dwóm rygorystycznym warunkom:
(i) tradycyjny odpowiednik zawsze występuje w tym samym bloku razem z odpowiednią rośliną zmodyfikowaną genetycznie;
(ii) wszystkie niezmodyfikowane genetycznie odmiany wzorcowe pojawiają się w każdym z niekompletnych bloków i są całkowicie zrandomizowane z roślinami i ich okazami porównawczymi.
Doświadczenia polowe są odpowiednio opisane z uwzględnieniem informacji na temat istotnych parametrów, takich jak: gospodarowanie polem przed siewem, data siewu, typ gleby, stosowanie herbicydów, warunki klimatyczne i inne warunki uprawy/środowiskowe podczas wzrostu roślin i w czasie zbioru, jak również warunki podczas przechowywania zebranego materiału.
Bardziej szczegółowe wytyczne dotyczące stosowania wymogów zawartych w niniejszej sekcji są dostępne w opinii EFSA pt. „Statistical considerations for the safety evaluation of GMOs” (Czynniki statystyczne dotyczące oceny bezpieczeństwa GMO) (3).
1.3.2.2. Analiza statystyczna
Analizę danych przedstawia się w przejrzystej formie, przy użyciu znormalizowanych jednostek naukowych. Dane nieprzetworzone i kod programowania wykorzystywane do celów analizy statystycznej podaje się w formie edytowalnej.
Konieczne może być przekształcenie danych w celu zapewnienia normalności i odpowiedniej skali, na której skutki statystyczne są addytywne. W przypadku wielu objaśnianych zmiennych punktów końcowych odpowiednia powinna być transformacja logarytmiczna. W takich przypadkach każda różnica między materiałem genetycznie zmodyfikowanym a jakimkolwiek innym materiałem badawczym jest interpretowana jako współczynnik w skali naturalnej. Gdy jednak transformacja logarytmiczna nie dostarcza właściwych wyników, bierze się pod uwagę skalę naturalną lub inną.
Całkowita zmienność każdego punktu końcowego obserwowana w doświadczeniach polowych jest oceniana i rozdzielana przy pomocy odpowiednich modeli statystycznych w celu uzyskania dwóch zbiorów granic ufności oraz określenia dolnej i górnej granicy równoważności w oparciu o zmienność obserwowaną wśród odmian wzorcowych. Jeden zbiór granic ufności jest stosowany w badaniu różnicy; drugi zbiór i granice równoważności są wykorzystywane w badaniu równoważności.
Do obliczania granic ufności na potrzeby obu badań (tj. badania różnicy i badania równoważności) stosuje się liniowy mieszany model statystyczny; nieco inny model stosuje się do oszacowania granic równoważności, które wykorzystuje się w badaniu równoważności.
Zmienną wskaźnikową (niecentrowaną w modelu mieszanym) należy oznaczyć literą I, w taki sposób, że I = 1 dla działki pola, na której znajduje się jakakolwiek z niezmodyfikowanych genetycznie odmian wzorcowych, natomiast w pozostałych przypadkach I = 0. Następnie czynnikami losowymi dla modelu 1 powinny być – lecz niekoniecznie ograniczać się do nich – czynniki reprezentujące zmienność: (i) między materiałami badawczymi (zestaw zawierający genetycznie zmodyfikowaną roślinę, jej tradycyjny odpowiednik, każdą z niezmodyfikowanych genetycznie odmian wzorcowych oraz wszelkie dodatkowe okazy porównawcze); (ii) w interakcji między materiałami badawczymi a I; (iii) między miejscami; oraz (iv) między blokami w obrębie miejsc. Model 2 powinien być identyczny jak model 1, z tą różnicą, że pomija się czynnik losowy reprezentujący interakcję miedzy materiałami badawczymi a I.
Czynnik stały dla obu modeli powinien mieć tyle poziomów, ile jest materiałów badawczych, i reprezentować kontrasty między średnimi materiałów badawczych. Materiały badawcze definiuje się tak jak powyżej: genetycznie zmodyfikowana roślina; jej tradycyjny odpowiednik; zbiór niezmodyfikowanych genetycznie odmian wzorcowych oraz wszelkie dodatkowe materiały badawcze. Zbiór niezmodyfikowanych genetycznie odmian wzorcowych uznaje się za pojedynczy poziom czynnika stałego. Na potrzeby badania różnicy elementem badanego czynnika stałego jest kontrast o pojedynczym stopniu swobody między genetycznie zmodyfikowaną rośliną a jej tradycyjnym odpowiednikiem. Na potrzeby badania równoważności elementem badanego czynnika stałego jest kontrast o pojedynczym stopniu swobody między genetycznie zmodyfikowaną rośliną a zbiorem niezmodyfikowanych genetycznie odmian wzorcowych.
Zarówno badanie różnicy, jak i badanie równoważności przeprowadza się przy pomocy zgodności między testowaniem hipotez a konstruowaniem granic ufności. W przypadku badania równoważności stosowane podejście jest zgodne z metodą dwóch jednostronnych testów (TOST), w ramach której odrzuca się zerową hipotezę nierównoważności, gdy obie granice ufności mieszczą się w granicach równoważności. Wybór granic ufności na poziomie 90 % odpowiada zwyczajowemu poziomowi 95 % w statystycznym badaniu równoważności.
Wyniki badań różnicy i równoważności są przedstawiane wizualnie jednocześnie dla wszystkich punktów końcowych, na jednym wykresie lub na kilku wykresach.
Na wykresach pokazuje się linię zerowej różnicy między materiałem genetycznie zmodyfikowanym a jego tradycyjnym odpowiednikiem oraz – dla każdego punktu końcowego – dolne i górne skorygowane granice równoważności, średnią różnicę między materiałem genetycznie zmodyfikowanym a jego tradycyjnym odpowiednikiem oraz granice ufności dla tej różnicy (zob. zbiór możliwych wyników dla pojedynczego punktu końcowego na wykresie na rysunku 1).
Kiedy oprócz tradycyjnego odpowiednika używa się innego materiału badawczego jako okazu porównawczego, średnią różnicę między materiałem genetycznie zmodyfikowanym a tym okazem, jej granice ufności i jej skorygowane granice równoważności przedstawia się na wykresach dla wszystkich takich dodatkowych okazów porównawczych, odnosząc dane do tej samej zerowej linii podstawowej co określona przez tradycyjny odpowiednik. Linia zerowej różnicy w skali logarytmicznej odpowiada multiplikatywnemu czynnikowi jedności w skali naturalnej. Na osi poziomej oznacza się wartości, które określają zmianę w skali naturalnej. W przypadku transformacji logarytmicznej zmiany 2x i ½x pojawią się równomiernie rozmieszczone po którejkolwiek stronie linii zerowej różnicy.
Pomimo spodziewanego odsetka fałszywie istotnych różnic wnioskodawca zgłasza i omawia wszystkie istotne różnice zaobserwowane między uprawą zmodyfikowaną genetycznie, jej tradycyjnym odpowiednikiem oraz, w stosownych przypadkach, każdym innym materiałem badawczym, skupiając się na ich znaczeniu biologicznym (zob. sekcja 3, która dotyczy charakterystyki ryzyka).
Dla celów sprawozdawczości podaje się pełne szczegóły dla każdego przeanalizowanego punktu końcowego, wymieniając:
a) założenia leżące u podstawy analizy;
b) pełną specyfikację wybranych modeli mieszanych, w tym z efektami stałymi i losowymi;
c) wyniki wszelkich badań wzajemnego oddziaływania między materiałami badawczymi a miejscami;
d) efekty stałe wraz z odpowiednią szacunkową zmiennością resztową, z którą są porównywane, i komponentami wariancyjnymi dla czynników losowych;
e) szacunkowe stopnie swobody;
f) wszelkie inne istotne dane statystyczne.
Przedstawia się omówienie prawdopodobnego wpływu innych warunków uprawy, których nie zbadano w doświadczeniu polowym.
Rysunek 1. Uproszczona wersja wykresu dla oceny porównawczej z przedstawieniem siedmiu typów wyników możliwych dla każdego pojedynczego punktu końcowego. Po skorygowaniu granic równoważności jedna granica ufności (dotycząca różnicy) służy jako pomoc wizualna do celów oceny wyniku obu badań (różnicy i równoważności). W omawianym przypadku bierze się pod uwagę tylko górną skorygowaną granicę równoważności. Na wykresie poniżej przedstawiono: średnią uprawy zmodyfikowanej genetycznie w odpowiedniej skali (kwadrat); granice ufności (wąsy wykresu) dla różnicy między uprawą zmodyfikowaną genetycznie a jej tradycyjnym odpowiednikiem (pasek pokazuje przedział ufności); pionową linię wskazującą brak różnicy (w odniesieniu do badania różnicy); i pionowe linie wskazujące skorygowane granice równoważności (w odniesieniu do badania równoważności). Dla typów wyników 1, 3 i 5 nie można odrzucić hipotezy zerowej o braku różnicy: w przypadku wyników 2, 4, 6 i 7 uprawa zmodyfikowana genetycznie różni się od swojego tradycyjnego odpowiednika. Jeśli chodzi o interpretację równoważności, określono cztery kategorie, tj. od (i) do (iv): w kategorii (i) hipotezę zerową o nierównoważności odrzuca się na rzecz równoważności; w kategorii (ii), (iii) oraz (iv) nie można odrzucić nierównoważności.
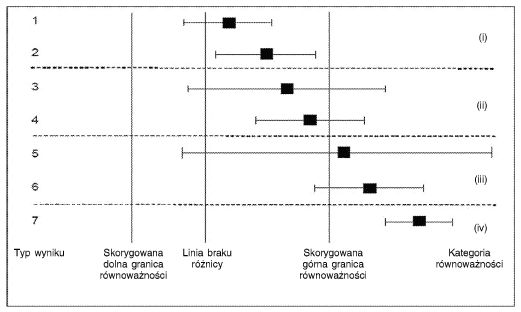
A. Jeśli chodzi o badanie różnicy, każdy wynik z wykresu klasyfikuje się w następujący sposób i odpowiednio wyciąga właściwe wnioski.
(i) Typy wyników 1, 3 i 5: pasek przedziału ufności częściowo pokrywa się z linią braku różnicy. Nie można odrzucić hipotezy zerowej o braku różnicy i właściwy wniosek jest taki, że nie ma wystarczających dowodów na to, że uprawa zmodyfikowana genetycznie i jej tradycyjny odpowiednik różnią się.
(ii) Typy wyników 2, 4, 6 i 7: pasek przedziału ufności nie pokrywa się z linią braku różnicy. Trzeba odrzucić hipotezę zerową o braku różnicy i właściwy wniosek jest taki, że uprawa zmodyfikowana genetycznie znacząco różni się od swojego tradycyjnego odpowiednika.
B. Jeśli chodzi o badanie równoważności, każdy wynik z wykresu klasyfikuje się w następujący sposób i odpowiednio wyciąga właściwe wnioski.
(i) Typy wyników 1 i 2 (kategoria (i), rysunek 1): obie granice ufności leżą między skorygowanymi granicami równoważności i odrzuca się hipotezę zerową o nierównoważności. Właściwy wniosek jest taki, że uprawa zmodyfikowana genetycznie jest równoważna zbiorowi niezmodyfikowanych genetycznie odmian wzorcowych.
(ii) Typy wyników 3 i 4 (kategoria (ii), rysunek 1): średnia uprawy zmodyfikowanej genetycznie leży między skorygowanymi granicami równoważności, ale pasek przedziału ufności częściowo pokrywa się na wykresie z co najmniej jedną skorygowaną granicą równoważności. Nie można odrzucić nierównoważności, a właściwy wniosek jest taki, że równoważność między uprawą zmodyfikowaną genetycznie a zbiorem niezmodyfikowanych genetycznie odmian wzorcowych jest bardziej prawdopodobna niż brak równoważności.
(iii) Typy wyników 5 i 6 (kategoria (iii), rysunek 1): średnia uprawy zmodyfikowanej genetycznie leży poza skorygowanymi granicami równoważności, ale pasek przedziału ufności częściowo pokrywa się z co najmniej jedną skorygowaną granicą równoważności. Nie można odrzucić nierównoważności, a właściwy wniosek jest taki, że brak równoważności między uprawą zmodyfikowaną genetycznie a zbiorem niezmodyfikowanych genetycznie odmian wzorcowych jest bardziej prawdopodobny niż równoważność.
(iv) Typ wyników 7 (kategoria (iv), rysunek 1): obie granice ufności leżą poza skorygowanymi granicami równoważności. Właściwy wniosek jest taki, że występuje brak równoważności między uprawą zmodyfikowaną genetycznie a zbiorem niezmodyfikowanych genetycznie odmian wzorcowych.
W przypadku znaczącej różnicy lub braku równoważności w odniesieniu do jakiegokolwiek konkretnego punktu końcowego przeprowadza się dalszą analizę statystyczną, aby ocenić, czy istnieje wzajemne oddziaływanie między materiałami badawczymi a miejscem. Można tego dokonać np. przy pomocy zwykłej standardowej metody analizy wariancji (ANOVA). Niezależnie od przyjętego podejścia podaje się szczegóły dla każdego przeanalizowanego punktu końcowego, wymieniając: a) założenia leżące u podstaw analizy; oraz, w stosownych przypadkach: b) stopnie swobody; c) szacunkową zmienność resztową dla każdego źródła zmienności i komponenty wariancyjne; (d) wszelkie inne istotne dane statystyczne. Te dodatkowe analizy mają pomóc w interpretacji wszelkich znaczących wykrytych różnic i w zbadaniu potencjalnego wzajemnego oddziaływania między materiałami badawczymi i innymi czynnikami.
Bardziej szczegółowe wytyczne dotyczące stosowania wymogów zawartych w niniejszej sekcji są dostępne w opinii EFSA pt. „Statistical considerations for the safety evaluation of GMOs” (Czynniki statystyczne dotyczące oceny bezpieczeństwa GMO) (4).
1.3.3. Wybór materiału i związków do analizy
Analiza składu materiału roślinnego ma zasadnicze znaczenie przy porównywaniu genetycznie zmodyfikowanej żywności i paszy z ich tradycyjnymi odpowiednikami. Materiał wykorzystywany dla celów oceny porównawczej wybiera się przy uwzględnieniu zastosowań rośliny zmodyfikowanej genetycznie i charakteru modyfikacji genetycznej. W przypadku roślin zmodyfikowanych genetycznie tolerujących herbicydy stosuje się trzy materiały badawcze: roślinę zmodyfikowaną genetycznie wystawioną na działanie zamierzonego herbicydu, tradycyjny odpowiednik objęty konwencjonalnymi systemami traktowania herbicydami oraz roślinę zmodyfikowaną genetycznie objętą tymi samymi konwencjonalnymi systemami traktowania herbicydami. O ile nie jest to należycie uzasadnione, analizę przeprowadza się na nieprzetworzonym towarze rolnym, ponieważ zazwyczaj stanowi on główny punkt wprowadzenia materiału do łańcucha produkcji i przetwarzania żywności i paszy. W poszczególnych przypadkach, w miarę potrzeby, jest przeprowadzana dodatkowa analiza produktów przetworzonych (np. żywności i paszy, składników żywności, materiałów paszowych, dodatków do żywności i dodatków paszowych lub środków aromatyzujących do żywności) (zob. sekcja 1.3.6). Pobieranie próbek, analizę i przygotowanie materiału badawczego przeprowadza się zgodnie z odpowiednimi standardami jakości.
1.3.4. Analiza porównawcza składu
Oprócz analizy na poziomie nowych białek ulegających ekspresji (zob. sekcja 1.2.2.3) analizę składu przeprowadza się w odniesieniu do odpowiednich związków. W każdym przypadku wnioskodawca dostarcza co najmniej analizę składu podstawowego (w tym wilgoci i popiołu całkowitego), najważniejszych makro- i mikroskładników pokarmowych, substancji obniżających właściwości odżywcze pokarmów, naturalnych toksyn i już zidentyfikowanych alergenów, jak również innych drugorzędnych metabolitów roślinnych charakterystycznych dla poszczególnych gatunków upraw, o których mowa w dokumentach porozumienia Organizacji Współpracy Gospodarczej i Rozwoju (OECD) dotyczących kwestii składu nowych odmian roślin (dokumenty porozumienia OECD) (5). Do analizy wybiera się te witaminy i składniki mineralne, które są obecne na poziomie istotnym dla żywienia lub które pod względem żywieniowym stanowią znaczący wkład w dietę na poziomie, na którym dana roślina jest spożywana. Szczegółowe wymagane analizy zależą od badanego gatunku rośliny, lecz obejmują szczegółową ocenę stosowną do zamierzonego skutku modyfikacji genetycznej, wartości odżywczej i zastosowania danej rośliny. Wnioskodawca zwraca szczególną uwagę na najważniejsze składniki odżywcze, takie jak: białka, węglowodany, lipidy/tłuszcze, błonnik, witaminy i składniki mineralne. Na przykład profil kwasów tłuszczowych bierze się pod uwagę w przypadku roślin bogatych w olej (główne poszczególne nasycone, jednonienasycone i wielonienasycone kwasy tłuszczowe), natomiast profil aminokwasowy (poszczególne aminokwasy białkowe i główne aminokwasy niebiałkowe) bada się w przypadku roślin wykorzystywanych jako ważne źródło białka. W przypadku wegetatywnych części roślin wykorzystywanych w paszy wymagana jest również analiza dotycząca składników ścian komórkowych roślin.
Wnioskodawca przedstawia również analizę dotyczącą najważniejszych toksyn z natury występujących w roślinie biorcy, które w zależności od ich mocy i stężenia mogą mieć szkodliwy wpływ na zdrowie ludzi/zwierząt. Stężenia tych związków ocenia się według gatunków roślin i proponowanego zastosowania żywności i paszy. Podobnie bada się substancje obniżające właściwości odżywcze pokarmów, takie jak inhibitory enzymów trawiennych, i już zidentyfikowane alergeny.
Właściwości wprowadzonej cechy mogą decydować o przeprowadzeniu dalszej analizy poszczególnych związków, w tym metabolitów potencjalnie zmodyfikowanych szlaków metabolicznych. W razie potrzeby wnioskodawca bierze pod uwagę włączenie związków innych niż najważniejsze składniki odżywcze, toksyny, substancje obniżające właściwości odżywcze pokarmów i alergeny wskazane w dokumentach porozumienia OECD oraz przedstawia uzasadnienie wyboru tych związków.
1.3.5. Analiza porównawcza właściwości agronomicznych i fenotypowych
Wnioskodawca przedstawia porównanie między rośliną zmodyfikowaną genetycznie a jej tradycyjnym odpowiednikiem. Porównanie to ma umożliwić wnioskodawcy określenie niezamierzonych skutków wynikających z modyfikacji genetycznej. Powinno uwzględniać również biologię rośliny i cechy agronomiczne, w tym wspólne parametry hodowlane (takie jak: plony, morfologia rośliny, czas kwitnienia, liczba stopniodni do osiągnięcia dojrzałości, żywotność pyłku, reakcję na patogeny i szkodniki owadzie roślin, wrażliwość na obciążenie abiotyczne). Protokoły z tych doświadczeń polowych odpowiadają specyfikacjom określonym w sekcji 1.3.2.
W przypadku łączenia modyfikacji genetycznych przez tradycyjne metody rozmnażania mogą również wystąpić zmiany właściwości agronomicznych i fenotypowych. Ewentualne różnice pod względem cech fenotypowych i właściwości agronomicznych złożonych modyfikacji genetycznych ocenia się w doświadczeniach polowych. W stosownych przypadkach wnioskodawca przedstawia dodatkowe informacje na temat agronomicznych cech złożonych modyfikacji genetycznych pochodzące z dodatkowych doświadczeń polowych.
1.3.6. Skutki przetwarzania
Wnioskodawca ocenia, czy zastosowane technologie przetwarzania lub konserwowania mogą powodować zmianę właściwości genetycznie zmodyfikowanych produktów końcowych w porównaniu z ich stosownym tradycyjnym odpowiednikiem. Wnioskodawca przedstawia wystarczająco szczegółowy opis różnych technologii przetwórczych, zwracając szczególną uwagę na kroki, które mogą prowadzić do istotnych zmian w składzie, jakości i czystości produktu.
Modyfikacja genetyczna może być ukierunkowana na szlaki metaboliczne, co powoduje zmiany stężenia substancji niebiałkowych lub powstawanie nowych metabolitów (np. w żywności o podwyższonych właściwościach odżywczych). Przetworzone produkty mogą być oceniane przy przeprowadzaniu oceny rośliny zmodyfikowanej genetycznie pod kątem bezpieczeństwa modyfikacji genetycznej lub też przetworzony produkt można ocenić osobno. Wnioskodawca przedstawia naukowe uzasadnienie dla oceny ryzyka tych produktów. W poszczególnych przypadkach wnioskodawca rozważa przedłożenie dodatkowych danych doświadczalnych.
W stosownych przypadkach, w zależności od produktu, niezbędne są informacje na temat składu, poziomu substancji niepożądanych, wartości odżywczej i metabolizmu, a także planowanego wykorzystania.
W stosownych przypadkach, w zależności od charakteru nowych białek ulegających ekspresji, niezbędne jest ocenienie stopnia, w jakim etapy przetwarzania prowadzą do koncentracji lub do eliminacji, denaturacji lub degradacji tych białek w produkcie końcowym.
1.3.7. Wniosek
We wniosku z analizy porównawczej jasno określa się:
a) czy właściwości agronomiczne i fenotypowe rośliny zmodyfikowanej genetycznie, z wyjątkiem cech wprowadzonych, różnią się od właściwości jej tradycyjnego odpowiednika lub są równoważne właściwościom odmian wzorcowych, biorąc pod uwagę naturalną zmienność;
b) czy właściwości składu genetycznie zmodyfikowanej żywności i paszy, z wyjątkiem wprowadzonych cech, różnią się od właściwości jej tradycyjnego odpowiednika lub są równoważne właściwościom odmian wzorcowych, biorąc pod uwagę naturalną zmienność;
c) właściwości, z powodu których roślina zmodyfikowana genetycznie lub genetycznie zmodyfikowana żywność i pasza różnią się od ich tradycyjnego odpowiednika lub nie są równoważne odmianom wzorcowym i które wymagają dalszego zbadania, biorąc pod uwagę naturalną zmienność;
d) czy w przypadku modyfikacji genetycznych połączonych wskutek konwencjonalnego krzyżowania istnieją oznaki wzajemnego oddziaływania między złożonych modyfikacjami genetycznymi.
1.4. Toksykologia
Ocenia się wpływ toksykologiczny wszelkich zmian na całą genetycznie zmodyfikowaną żywność lub paszę, wynikających z modyfikacji genetycznej, takiej jak wprowadzenie nowych genów, wyciszenie genu lub nadmierna ekspresja genu endogenicznego.
Ocenę toksykologiczną przeprowadza się w celu:
a) wykazania, że zamierzone skutki modyfikacji genetycznej nie mają szkodliwego wpływu na zdrowie ludzi i zwierząt;
b) wykazania, że niezamierzone skutki modyfikacji genetycznych, których wystąpienie stwierdzono lub założono w oparciu o wcześniejsze porównawcze analizy molekularne, składu lub fenotypowe, nie mają szkodliwego wpływu na zdrowie ludzi i zwierząt;
c) określenia potencjalnych szkodliwych skutków nowych składników i ustalenia ich najwyższej dawki, która nie powoduje szkodliwych skutków. Dopuszczalne dzienne spożycie pojedynczych związków przez ludzi można uzyskać na podstawie danych pochodzących z odpowiedniego badania na zwierzętach, poprzez zastosowanie współczynników niepewności lub bezpieczeństwa, które uwzględniają różnice pomiędzy gatunkiem badanego zwierzęcia a człowiekiem oraz osobniczą zmienność między ludźmi;
d) określenia potencjalnych szkodliwych skutków dla całej genetycznie zmodyfikowanej żywności lub paszy bądź wyeliminowania pozostających wątpliwości dzięki przeprowadzeniu 90-dniowych badań żywieniowych.
Wnioskodawca rozważa charakter badań toksykologicznych, które mają być przeprowadzone na nowych składnikach i na całej genetycznie zmodyfikowanej żywności lub paszy, w oparciu o wynik analiz molekularnej i porównawczej, o których mowa w sekcjach 1.2 i 1.3, a mianowicie o różnice stwierdzone pomiędzy produktem zmodyfikowanym genetycznie a jego tradycyjnym odpowiednikiem, w tym zmiany zarówno zamierzone, jak i niezamierzone. Wnioskodawca ocenia również wyniki wykonanych badań toksykologicznych, aby rozważyć potrzebę przeprowadzenia dodatkowych badań na nowych składnikach lub na całej genetycznie zmodyfikowanej żywności lub paszy zgodnie z sekcjami 1.4.4.2 i 1.4.4.3.
Wnioskodawca uwzględnia obecność: nowych białek ulegających ekspresji, potencjalną obecność innych nowych składników lub możliwe zmiany w poziomie naturalnych składników wykraczające poza normalną zmienność. Szczegółowe wymagania dotyczące informacji i strategie badań przedstawiono w sekcjach 1.4.1 do 1.4.4.
Jeśli chodzi o wnioski, których zakres obejmuje genetycznie zmodyfikowaną żywność i paszę produkowaną z roślin zmodyfikowanych genetycznie lub jest do nich ograniczony, przeprowadza się badania toksykologiczne produktów przetworzonych, z wyjątkiem przypadków, w których wnioskodawca przedstawi ocenę ryzyka w odniesieniu do rośliny zmodyfikowanej genetycznie (lub odpowiednich jej części), wykazującą jej bezpieczeństwo, i gdy nie ma przesłanek za tym, że przetworzona genetycznie zmodyfikowana żywność i pasza w jakiś sposób różni się od swojego właściwego tradycyjnego odpowiednika. W tej kwestii wnioskodawca przedstawia odpowiednie uzasadnienie.
Badania toksykologiczne mające na celu ocenę ryzyka dla zdrowia ludzi lub zwierząt wzajemnie się uzupełniają. Większość badań niezbędnych do oceny bezpieczeństwa genetycznie zmodyfikowanej żywności zachowuje także ważność w odniesieniu do oceny genetycznie zmodyfikowanej paszy.
Oprócz narażenia konsumentów i zwierząt poprzez spożycie żywności i paszy wnioskodawca zgłasza wszelkie szkodliwe skutki dla osób, które mogą być spowodowane narażeniem ich na genetycznie zmodyfikowaną żywność i paszę w ramach obowiązków zawodowych, na przykład w rolnictwie, przy obróbce nasion. Przeprowadza się odpowiednie badania w celu dalszego scharakteryzowania tych oznak potencjalnych szkodliwych skutków.
W odniesieniu do badania toksyczności wnioskodawca stosuje przyjęte na szczeblu międzynarodowym protokoły i metody badań (zob. tabele 1 i 2 w sekcji 1.7). Modyfikacje tych protokołów lub zastosowanie ewentualnych innych metod odbiegających od nich uzasadnia się we wniosku.
1.4.1. Badanie nowych białek ulegających ekspresji
Wnioskodawca przedstawia ocenę wszystkich nowych białek ulegających ekspresji. Badania wymagane do zbadania potencjalnej toksyczności nowego białka ulegającego ekspresji wybierane są oddzielnie dla każdego przypadku, w zależności od dostępnej wiedzy na temat źródła tego białka, jego funkcji lub działania oraz historii spożycia przez ludzi lub zwierzęta. Jeśli chodzi o białka ulegające ekspresji w roślinie zmodyfikowanej genetycznie, w przypadku gdy istnieje należycie udokumentowana historia bezpiecznego stosowania jako żywności lub paszy zarówno rośliny, jak i nowego białka ulegającego ekspresji, specjalne badanie toksyczności przewidziane w niniejszej sekcji nie jest wymagane. W takim przypadku wnioskodawca podaje niezbędne informacje dotyczące historii bezpiecznego stosowania danych białek.
Jeżeli wymagane jest specjalne badanie, badane białko jest równoważne nowemu białku ulegającemu ekspresji w roślinie zmodyfikowanej genetycznie. Jeżeli z powodu braku wystarczającej ilości badanego materiału z rośliny wykorzystywane jest białko wytwarzane przez mikroorganizmy, wykazuje się równoważność strukturalną, biochemiczną i funkcjonalną tego mikrobiologicznego substytutu z nowym białkiem roślinnym ulegającym ekspresji. Do wykazania równoważności niezbędne jest w szczególności porównanie masy cząsteczkowej, sekwencji aminokwasów, modyfikacji potranslacyjnej, reaktywności immunologicznej oraz, w przypadku enzymów, aktywności enzymatycznej. W przypadku różnic pomiędzy białkiem roślinnym ulegającym ekspresji a jego mikrobiologicznym substytutem ocenia się znaczenie tych różnic dla badań bezpieczeństwa.
Aby wykazać bezpieczeństwo nowego białka ulegającego ekspresji, wnioskodawca przedstawia:
a) molekularną i biochemiczną charakterystykę nowego białka ulegającego ekspresji, w tym określenie struktury podstawowej, masy cząsteczkowej (np. przy pomocy spektrometrii masowej), badania nad modyfikacjami potranslacyjnymi i opis jego funkcji. W przypadku nowych enzymów ulegających ekspresji podaje się również informacje na temat działania enzymu, w tym zakresu temperatury i pH dla optymalnego działania enzymu, specyficzności substratowej i możliwych produktów reakcji. Ocenia się również potencjalne wzajemne oddziaływanie z innymi składnikami rośliny;
b) aktualne wyniki wyszukiwania homologii z białkami, co do których wiadomo, że wywołują szkodliwe skutki, takimi jak białka toksyczne. Wyszukiwanie homologii z białkami wpływającymi na normalne funkcje metaboliczne lub strukturalne również może dostarczyć cennych informacji. Określa się bazy danych i metodykę wykorzystane do przeprowadzenia wyszukiwania;
c) opis stabilności białka w odpowiednich warunkach przetwarzania i przechowywania, a także przewidywanej obróbki żywności i paszy. Bada się wpływ zmian temperatury i pH oraz charakteryzuje się potencjalne zmiany białek (takie jak denaturacja) lub produkcję stabilnych fragmentów białek wytwarzanych wskutek takiego przetwarzania.
d) dane dotyczące oporności nowych białek ulegających ekspresji na enzymy proteolityczne (np. pepsynę), takie jak uzyskiwane w drodze badań in vitro z wykorzystaniem odpowiednich i znormalizowanych testów. Stabilne produkty rozkładu są charakteryzowane i oceniane w odniesieniu do możliwości wywierania szkodliwego wpływu na zdrowie związanego z aktywnością biologiczną tych produktów;
e) badanie pokarmowej toksyczności dla dawki powtarzalnej nowego białka ulegającego ekspresji, prowadzane przez 28 dni na gryzoniach. W stosownych przypadkach, w zależności od wyniku 28-dniowego badania toksyczności, przeprowadzane są dalsze ukierunkowane badania, w tym analiza immunotoksyczności.
Badanie toksyczności ostrej nowych białek ulegających ekspresji pochodzących z roślin zmodyfikowanych genetycznie ma niewielką wartość dodatkową dla oceny ryzyka dotyczącej powtarzanego spożywania przez ludzi i zwierzęta genetycznie zmodyfikowanej żywności i paszy i nie jest dostarczane jako część badań przeprowadzanych w ramach niniejszego punktu.
Jeżeli wynikiem modyfikacji genetycznej jest ekspresja dwóch lub większej liczby białek w roślinie zmodyfikowanej genetycznie i gdy w oparciu o wiedzę naukową stwierdza się potencjalne synergiczne lub antagonistyczne wzajemne oddziaływanie budzące obawy co do bezpieczeństwa, wnioskodawca przeprowadza badania, w ramach których białka podaje się łącznie.
1.4.2. Badanie nowych składników innych niż białka
Wnioskodawca przedstawia ocenę ryzyka określonych nowych składników innych niż białka. Obejmuje to, oddzielnie dla każdego przypadku, ocenę ich potencjału toksycznego i potrzebę przeprowadzenia badań toksykologicznych, a także określenie ich stężenia w genetycznie zmodyfikowanej żywności i paszy. Aby ustalić bezpieczeństwo nowych składników, które nie mają historii bezpiecznego stosowania w żywności i paszy przeznaczonych do spożycia, wnioskodawca podaje informacje analogiczne do informacji opisanych w „Guidance for submissions for food additive evaluations by the EFSA Panel on Food Additives and Nutrient Sources added to Food” (Wytycznych dla składania wniosków o ocenę dodatków do żywności przez Panel EFSA ds. dodatków do żywności i składników pokarmowych dodawanych do żywności) z dnia 16 sierpnia 2012 r. (6) i w rozporządzeniu Komisji (WE) nr 429/2008 z dnia 25 kwietnia 2008 r. w sprawie szczegółowych zasad wykonania rozporządzenia (WE) nr 1831/2003 w zakresie sporządzania i przedstawiania wniosków oraz oceny dodatków paszowych i udzielania zezwoleń na dodatki paszowe (7). Obejmuje to przedstawienie informacji na temat podstawowego zbioru badań, takich jak badania metabolizmu/toksykokinetyki, toksyczności podprzewlekłej, genotoksyczności, toksyczności przewlekłej, działania rakotwórczego oraz toksyczności reprodukcyjnej i rozwojowej, wraz z dowolnymi innymi stosownymi rodzajami badań. Szczegółowe wytyczne dotyczące badań na zwierzętach znajdują się w tabeli 1 w sekcji 1.7 niniejszego załącznika. Protokoły badań genotoksyczności są przedstawione w tabeli 2 w sekcji 1.7 niniejszego załącznika.
1.4.3. Informacje na temat zmienionych poziomów składników żywności i paszy
Niniejszą sekcję stosuje się wyłącznie w przypadku, gdy zamierzony lub niezamierzony skutek modyfikacji genetycznej powodowałby zmianę poziomów składników żywności i paszy wykraczającą poza ich naturalną zmienność.
Aby wykazać bezpieczeństwo zmienionych poziomów składników żywności i paszy, takich jak: makro- i mikroskładniki pokarmowe, substancje obniżające właściwości odżywcze pokarmów i naturalne toksyny, a także inne wtórne metabolity roślinne, wnioskodawca przedstawia szczegółową ocenę ryzyka w oparciu o wiedzę o funkcji fizjologicznej lub właściwościach toksycznych tych składników.
Wynik tej oceny ryzyka decyduje o tym, czy i w jakim zakresie wnioskodawca przeprowadza dodatkowe badania toksykologiczne wybranych składników żywności i paszy, oprócz 90-dniowych badań żywieniowych na gryzoniach karmionych całą genetycznie zmodyfikowaną żywnością lub paszą.
1.4.4. Badanie przeprowadzone na całej genetycznie zmodyfikowanej żywności i paszy
Swoją ocenę ryzyka dotyczącą genetycznie zmodyfikowanej żywności i paszy wnioskodawca opiera głównie na charakterystyce molekularnej, porównawczej analizie agronomicznej i fenotypowej oraz porównawczej kompleksowej analizie składu, a także na toksykologicznej ocenie określonych zamierzonych i niezamierzonych skutków, w tym na 90-dniowych badaniach żywieniowych na gryzoniach karmionych całą genetycznie zmodyfikowaną żywnością lub paszą, przeprowadzanych zgodnie z opisem w sekcji 1.4.4.1. W okolicznościach określonych w pkt 1.4.4.2 i 1.4.4.3 niniejszej sekcji wykonuje się dodatkowe specyficzne badania toksykologiczne na całej genetycznie zmodyfikowanej żywności i paszy.
1.4.4.1. 90-dniowe badania żywieniowe na gryzoniach karmionych całą genetycznie zmodyfikowaną żywnością lub paszą
Wnioskodawca dołącza do wniosku 90-dniowe badania żywieniowe na gryzoniach karmionych całą genetycznie zmodyfikowaną żywnością lub paszą na potrzeby oceny żywności i paszy zawierających rośliny zmodyfikowane genetycznie, składających się z nich lub wyprodukowanych z takich roślin; rośliny te zawierają pojedyncze modyfikacje genetyczne lub złożone modyfikacje genetyczne, których nie uzyskano poprzez konwencjonalne krzyżowanie roślin zmodyfikowanych genetycznie zawierających pojedyncze modyfikacje genetyczne.
W przypadku złożonych modyfikacji genetycznych uzyskanych poprzez konwencjonalne krzyżowanie roślin zmodyfikowanych genetycznie zawierających jedną lub kilka modyfikacji genetycznych 90-dniowe badanie żywieniowe na gryzoniach karmionych całą genetycznie zmodyfikowaną żywnością lub paszą przeprowadzane jest w odniesieniu do każdej wykorzystanej rośliny zmodyfikowanej genetycznie zawierającej pojedynczą modyfikację genetyczną. Dodatkowe 90-dniowe badania żywieniowe na gryzoniach karmionych całą genetycznie zmodyfikowaną żywnością lub paszą uzyskaną z rośliny zmodyfikowanej genetycznie ze złożonymi modyfikacjami genetycznymi przeprowadza się, jeżeli w trakcie oceny (i) stabilności insertów; (ii) ekspresji insertów; oraz (iii) potencjalnych efektów synergicznych lub antagonistycznych wynikających z kombinacji modyfikacji genetycznych wykryte zostaną oznaki potencjalnych szkodliwych skutków.
Program badania toksyczności z wykorzystaniem genetycznie zmodyfikowanej żywności i paszy należy wykonać zgodnie z „badaniem toksyczności podprzewlekłej drogą pokarmową – studium toksyczności na gryzoniach metodą powtarzanej 90-dniowej dawki drogą pokarmową” (zob. tabela 1), stosując przyjęty protokół. Co do zasady stosuje się co najmniej dwie dawki badawcze i kontrolę ujemną. Najwyższą dawką jest maksymalna dawka, jaką można uzyskać, nie powodując dysproporcji pod względem wartości odżywczych; najniższa dawka zawiera badaną żywność lub paszę w ilości zawsze powyżej przewidywanego poziomu spożycia przez ludzi lub zwierzęta, dla których pasza jest przeznaczona. Analizowana genetycznie zmodyfikowana żywność i pasza powinna mieć związek z produktem, który ma być spożywany. W przypadku genetycznie zmodyfikowanych roślin tolerujących herbicydy badany materiał powinien pochodzić z genetycznie zmodyfikowanej rośliny wystawionej na działanie przeznaczonego dla niej herbicydu. Informacje o naturalnej zmienności parametrów badawczych uzyskuje się w miarę możliwości z danych historycznych, a nie z włączenia do eksperymentów odmian wzorcowych, składających się z dostępnej w handlu żywności i paszy otrzymanej z niezmodyfikowanych genetycznie roślin posiadających historię bezpiecznego stosowania. Analiza statystyczna koncentruje się na wykrywaniu ewentualnych różnic między materiałem badawczym a jego próbką kontrolną. W celu oszacowania wielkości próbki umożliwiającej wykrycie skali wcześniej określonych skutków istotnych biologicznie należy stosować analizę mocy z zastosowaniem określonego poziomu mocy i istotności. Bardziej szczegółowe wskazówki dotyczące wykonywania tego badania przedstawiono w wytycznych EFSA w sprawie przeprowadzania metodą powtarzanej 90-dniowej dawki podawanej drogą pokarmową badania toksyczności na gryzoniach karmionych całą żywnością lub paszą (8).
1.4.4.2. Badania na zwierzętach w odniesieniu do szkodliwego wpływu na rozrodczość oraz do toksyczności rozwojowej
Jeżeli informacje wymagane w sekcjach 1.4.1, 1.4.2 i 1.4.3 w odniesieniu do genetycznie zmodyfikowanej żywności i paszy wskazują na potencjalny szkodliwy wpływ na rozrodczość, potencjalną toksyczność rozwojową lub przewlekłą, lub jeżeli 90-dniowe badania żywieniowe na gryzoniach wskazują na potencjalny szkodliwy wpływ (taki jak funkcjonalne lub histologiczne zmiany organów/tkanek nerwowych, dokrewnych, rozrodczych lub immunologicznych), przeprowadza się odpowiednie badania. Protokoły w sprawie badania szkodliwego wpływu na rozrodczość, toksyczności rozwojowej i przewlekłej (zob. tabela 1 w sekcji 1.7) mogą być dostosowane do celów badania całej genetycznie zmodyfikowanej żywności lub paszy.
Z uwagi na fakt, że 90-dniowe badania żywieniowe na gryzoniach służą wyłącznie do wykrywania skutków dla wagi i histopatologii organów rozrodczych osobników dorosłych i nie wykrywa się w nich innych skutków dla rozrodczości i rozwoju – jeżeli stwierdzono zagrożenia w tym zakresie, oprócz 90-dniowych badań żywieniowych na gryzoniach przeprowadza się badanie na całej żywności lub paszy.
1.4.4.3. Inne badania na zwierzętach w celu ocenienia bezpieczeństwa i cech genetycznie zmodyfikowanej żywności i paszy (zob. także sekcje 1.6.1 i 1.6.2)
Do wniosku dołącza się badania żywieniowe docelowego gatunku zwierząt, jeżeli informacje wymagane w sekcjach 1.4.1, 1.4.2 i 1.4.3 w odniesieniu do genetycznie zmodyfikowanej żywności lub paszy lub wyniki 90-dniowych badań żywieniowych na gryzoniach wskazują na szkodliwy wpływ. Badania te dotyczą przede wszystkim bezpieczeństwa nowych składników (nowych białek ulegających ekspresji i innych nowych składników), określania i charakterystyki niezamierzonych skutków i żywieniowych skutków wszelkich zamierzonych, zasadniczych zmian składu rośliny zmodyfikowanej genetycznie (zob. także sekcja 1.6).
Tego rodzaju badania są ograniczone do materiałów roślinnych nadających się do włączenia do diety i które pod względem wartości odżywczej można dostosować do odpowiedniej diety kontrolnej.
1.4.4.4. Interpretacja znaczenia badań na zwierzętach
Ocenia się odpowiednie skutki zaobserwowane w czasie badań na zwierzętach, aby określić potencjalne konsekwencje dla zdrowia ludzi i zwierząt oraz ocenić ich znaczenie dla bezpieczeństwa genetycznie zmodyfikowanej żywności i paszy. Ocenę tę można uzupełnić dodatkowymi informacjami i uwagami. Należy zwracać uwagę na to, że niektóre skutki mogą być specyficzne dla badanych zwierząt, lecz nie dla ludzi – ze względu na różnice międzygatunkowe.
Wnioskodawca w szczególności rozważa zależność dawka-odpowiedź przy parametrach, które uległy zmianie (tj. następuje proporcjonalny wzrost zmian przy zwiększonych dawkach), ponieważ w dużym stopniu świadczy ona o skutkach badanego składnika. Jeżeli różnica jest zauważalna wyłącznie przy zastosowaniu najwyższej dawki, rozważa się inne czynniki, aby ustalić, czy istnieje związek z podaniem dawki. Wnioskodawca może uzyskać informacje na temat zmienności tła przy danym parametrze na podstawie danych dotyczących innych zwierząt tego samego gatunku/szczepu, badanych w ramach tego samego lub innego doświadczenia, lub ze zharmonizowanych na szczeblu międzynarodowym baz danych.
W badaniach, w których wykorzystuje się zwierzęta obojga płci, zmiany zachodzące wyłącznie u zwierząt jednej płci wciąż mogą być ważnym wskaźnikiem skutku, w zależności od zmienianego parametru i mechanizmu, który mógł spowodować zmianę. Na przykład zwierzęta określonej płci mogą być bardziej lub nawet szczególnie podatne na zmiany spowodowane przez dany składnik niż zwierzęta innej płci, jak ma to miejsce w przypadku skutków hormonalnych.
Wnioskodawca określa również możliwe wzajemne relacje pomiędzy obserwowanymi zmianami pojedynczych parametrów, które mogą stanowić silniejszą przesłankę wystąpienia skutku. Na przykład uszkodzenie wątroby, które można zaobserwować w samej wątrobie jako zmianę obrazu histopatologicznego, obrazu makroskopowego i masy organu, może również wynikać ze zmienionych poziomów wydzielania niektórych związków produkowanych przez wątrobę, takich jak enzymy lub bilirubina w surowicy.
W odniesieniu do potencjalnej przyczyny obserwowanego skutku bierze się pod uwagę prawdopodobieństwo związku przyczynowego nie tylko dla badanego związku, ale także dla innych czynników, które również mogą mieć wpływ na wyniki (takich jak spadek masy ciała ze względu na zmniejszone spożywanie mniej smacznego pożywienia). Dodatkowe dane dla hipotezy dotyczącej związku przyczynowego pomiędzy badanym związkiem a skutkami dla badanych zwierząt mogą na przykład obejmować prognozowane dane dotyczące prawdopodobnych skutków pochodzące z doświadczeń in vitro i in silico oraz zależności dawka-odpowiedź zaobserwowane w trakcie badań na zwierzętach.
1.4.5. Wnioski z oceny toksykologicznej
Wnioski z oceny toksykologicznej wskazują na to, czy:
a) potencjalne szkodliwe skutki określone w innych częściach oceny bezpieczeństwa zostały potwierdzone, czy odrzucone;
b) dostępne informacje na temat nowych białek ulegających ekspresji i innych nowych składników powstałych w drodze modyfikacji genetycznej wskazują w szczególności na potencjalne szkodliwe skutki oraz czy i przy jakiej dawce stwierdzono szkodliwe skutki w określonych badaniach;
c) informacje na temat naturalnych składników, których poziomy różnią się od tych w tradycyjnych odpowiednikach, wskazują na potencjalne szkodliwe skutki, w szczególności czy i przy jakiej dawce stwierdzono szkodliwe skutki w określonych badaniach;
d) stwierdzono szkodliwe skutki w oparciu o badania przeprowadzone na całej genetycznie zmodyfikowanej żywności i paszy oraz przy jakiej dawce.
Wnioskodawca określa wynik oceny toksykologicznej w świetle przewidywanego spożycia genetycznie zmodyfikowanej żywności i paszy (zob. sekcja 2).
1.5. Alergenność
Alergia pokarmowa jest niepożądaną reakcją na żywność i stanowi istotny problem dla zdrowia publicznego. Alergia pokarmowa różni się od reakcji toksycznych i nietolerancji. Alergia jest patologiczną reakcją immunologiczną na konkretną substancję występującą jedynie u niektórych osób, w przypadku których połączone oddziaływanie zmian w środowisku i predyspozycja genetyczna doprowadziły do uwrażliwienia.
U osób cierpiących na alergie czasem nawet drobna ilość żywności, którą większość ludzi toleruje dobrze, może wywołać poważne objawy, a nawet zgon. Negatywne skutki dla zdrowia wywołuje nie sam alergen, ale nieprawidłowa reakcja osoby cierpiącej na alergię na ten alergen.
Alergię pokarmową mogą wywołać różne mechanizmy immunologiczne. Główną formą alergii pokarmowej jest jednak alergia pokarmowa IgE-zależna, która wywołuje najsilniejsze reakcje i jest jedyną formą powodującą reakcje zagrażające życiu. Na alergii pokarmowej IgE-zależnej skoncentrowano się w ocenie ryzyka alergenności GMO. Co ważne, przebieg alergii pokarmowej składa się z dwóch odrębnych faz: pierwsza faza to uwrażliwienie, podczas której nie występują żadne objawy, zaś zdolność systemu immunologicznego do reagowania wzrasta radykalnie. Następna faza to wywołanie reakcji (prowokacja) z objawami klinicznymi.
Po spożyciu alergen lub alergeny, czyli żywność lub składnik żywności o właściwościach uczulających, są do pewnego stopnia rozkładane przez enzymy trawienne, wchłaniane przez błonę śluzową jelit (niewielkie ich ilości nawet przez błonę śluzową jamy ustnej), przetwarzane przez wyspecjalizowane komórki systemu immunologicznego, a następnie kierowane do reaktywnych komórek immunologicznych, które są odpowiedzialne za reakcję immunologiczną. Uwrażliwienie może zajść również w przypadku kontaktu alergenu pokarmowego ze skórą lub gdy dostanie się on do organizmu poprzez drogi oddechowe.
Składniki, które odpowiadają za alergenność tak żywności, jak i pyłków, to większości białka. Niektóre produkty rozpadu białek, czyli fragmenty peptydów, mogą zachować część alergenności białka natywnego i w związku z tym również można je uznać za alergeny.
Szczególne ryzyko wystąpienia alergii w przypadku GMO wiąże się z: (i) kontaktem z nowym białkiem lub białkami ulegającymi ekspresji, które mogą pojawić się w jadalnych częściach roślin lub w pyłku; ten punkt związany jest z biologicznym źródłem transgenu; oraz (ii) zmianami w alergenności całej rośliny i jej produktów, na przykład z powodu nadmiernej ekspresji naturalnych alergenów endogennych będącej niezamierzonym efektem modyfikacji genetycznej; ten punkt związany jest z biologią samej rośliny biorcy.
Bardziej szczegółowe wytyczne dotyczące stosowania wymogów zawartych w niniejszej sekcji są dostępne w naukowej opinii EFSA na temat oceny alergenności zmodyfikowanych genetycznie roślin i mikroorganizmów oraz pochodnej żywności i paszy, zatwierdzonej dnia 30 czerwca 2010 r. (9).
1.5.1. Ocena alergenności nowych białek ulegających ekspresji
Alergenność nie jest swoistą, w pełni przewidywalną właściwością danego białka, lecz stanowi aktywność biologiczną wymagającą interakcji z osobami o danych predyspozycjach genetycznych. Alergenność zależy więc od różnorodności i zmienności genetycznej u osób z atopią. Częstotliwość, siła i specyficzność reakcji alergicznych zależy również od czynników geograficznych i środowiskowych. Z powodu takiego braku całkowitej przewidywalności konieczne jest uwzględnienie w ocenie alergenności kilku aspektów w celu uzyskania zbioru dowodów, które zminimalizują wszelką niepewność co do danego białka lub białek.
Podczas badania strukturalnej charakterystyki oraz biologicznych i fizykochemicznych właściwości nowych białek ulegających ekspresji konieczne jest, by badane białko było równoważne pod względem struktury i aktywności z nowym białkiem ulegającym ekspresji w roślinie zmodyfikowanej genetycznie. Badania przeprowadzone z wykorzystaniem oczyszczonych białek docelowych przygotowanych w drodze ekspresji w organizmach takich, jak Escherichia coli są akceptowalne, pod warunkiem że właściwości zastępczego białka mikrobiologicznego są identyczne z właściwościami białka ulegającego ekspresji w danej roślinie, a tym samym uwzględniają wszystkie modyfikacje potranslacyjne, które zaszły w tej roślinie.
Wnioskodawca sprawdza, czy źródło transgenu jest alergenne. Jeśli wprowadzany materiały genetyczny uzyskuje się z pszenicy, żyta, jęczmienia, owsa lub podobnych ziaren zbóż, wnioskodawca dokonuje również oceny nowych białek ulegających ekspresji pod względem ich potencjalnej roli w wywołaniu enteropatii z nadwrażliwością na gluten lub innych enteropatii, które nie są IgE-zależne. W razie łączenia modyfikacji genetycznych wnioskodawca przedstawia ocenę zwiększonego potencjału alergennego w odniesieniu do ludzi i zwierząt dla poszczególnych przypadków. Te potencjalne efekty mogą wynikać z addytywnego, synergicznego lub antagonistycznego działania produktów genowych.
Wnioskodawca stosuje w ocenie potencjału alergennego nowych białek ulegających ekspresji zintegrowane podejście oparte na analizie poszczególnych przypadków, czyli podejście oparte na analizie ciężaru dowodów. Takie podejście obejmuje:
a) Porównanie homologii sekwencji aminokwasów nowych białek ulegających ekspresji i znanych alergenów
W każdym przypadku poszukiwanie homologii sekwencji lub strukturalnych podobieństw między białkami ulegającymi ekspresji a znanymi alergenami przeprowadza się w celu zidentyfikowania potencjalnej reaktywności krzyżowej z IgE pomiędzy nowymi białkami a znanymi alergenami. Wnioskodawca zapewnia jakość i kompleksowość baz danych zgodną z aktualnym stanem wiedzy. Za minimalny wymóg uznaje się kryterium dopasowania zakładające identyczność ze znanym alergenem na poziomie 35 % w ramach sekwencji o długości co najmniej 80 aminokwasów. Należy podać wszystkie parametry dopasowania sekwencji zastosowane w analizie, włącznie z obliczeniem procentu identyczności (PID). Obliczenia PID dokonuje się w ramach sekwencji 80 aminokwasów z lukami, tak aby wstawione luki były traktowane jako niedopasowanie. W niektórych przypadkach w celu oceny krótkich fragmentów peptydowych, takich jak ORF, można przeprowadzić poszukiwanie sekwencji przyległych reszt aminokwasowych identycznych lub podobnych pod względem chemicznym. Poszukiwania tego jednak nie można przeprowadzać rutynowo w celu identyfikacji potencjalnych liniowych epitopów IgE-wiążących ze względu na jego niską czułość lub specyficzność.
b) Specjalne badanie przesiewowe z użyciem surowicy
Tam, gdzie wykryto przesłanki dla homologii sekwencji lub podobieństwa strukturalnego, istotna procedura oceny potencjału do wywołania reakcji alergicznej na skutek kontaktu z nowym białkiem ulegającym ekspresji u osób już uczulonych na krzyżowo reaktywne białka opiera się na badaniach in vitro, które mierzą zdolność specyficznej IgE z surowicy pacjentów cierpiących na alergię do wiązania testowego białka lub białek. Specyficzność i powinowactwo reakcji na IgE różnią się w zależności od osoby. W szczególności specyficzność przeciwciał IgE wobec różnych alergenów obecnych w danej żywności/źródle lub różnych epitopach obecnych w danym białku może różnić się między osobami cierpiącymi na alergię. W celu zoptymalizowania czułości badania należy wykorzystać indywidualne surowice osób z dobrze scharakteryzowaną alergią. Wnioskodawca przeprowadza specjalne badanie przesiewowe z użyciem surowicy w następujących przypadkach:
(i) jeśli źródło wprowadzonego genu uważa się za alergenne, nawet jeśli nie wykazano homologii sekwencji nowych białek ulegających ekspresji ze znanym alergenem; lub
(ii) jeśli źródła nie uważa się za alergenne, ale istnieją przesłanki relacji między nowymi białkami ulegającymi ekspresji a znanym alergenem, na podstawie homologii sekwencji lub podobieństwa struktury.
Specjalne badanie przesiewowe z użyciem surowicy przeprowadza się z wykorzystaniem indywidualnych surowic od osób z udowodnioną i dobrze scharakteryzowaną alergią na źródło lub na potencjalnie krzyżowo reagujący alergen za pomocą odpowiednich testów immunochemicznych. Odpowiednimi metodami są testy IgE-wiążące (takie jak test radioalergosorpcji (RAST) lub enzymatyczny test alergosorpcji (EAST)), test immunoenzymatyczny (ELISA) oraz elektroforeza wraz z techniką Western blot ze specyficznymi surowicami zawierającymi IgE).
c) Badanie odporności na pepsynę oraz badania strawności in vitro
Odporność na trawienie enzymami proteolitycznymi od dawna była uważana za charakterystyczną właściwość białek alergennych. Pomimo stwierdzenia braku bezwzględnej korelacji odporność białka na trawienie przez pepsynę stanowi dodatkowe kryterium, które należy uwzględnić w podejściu do oceny alergenności opartym na analizie ciężaru dowodów. Badanie odporności na pepsynę przeprowadza się zazwyczaj w warunkach raczej zestandaryzowanych, przy niskim pH i wysokim stosunku stężeń pepsyny do białka. Uznaje się przy tym, że badanie odporności na pepsynę nie odzwierciedla fizjologicznych warunków, w jakich zachodzi trawienie. Strawność nowych białek ulegających ekspresji w szczególnych segmentach populacji, takich jak niemowlęta oraz osoby z upośledzonym funkcjonowaniem układu trawiennego, można oceniać za pomocą badań strawności in vitro, stosując inne warunki. Ponadto, ponieważ białko kodowane przez nowo wprowadzone geny będzie obecne w produkcie jako element złożonego materiału, należy uwzględnić w dodatkowych badaniach strawności in vitro wpływ możliwych oddziaływań pomiędzy białkiem a innymi składnikami mieszaniny, jak również skutki jej obróbki. W zależności od wyniku badania strawności in vitro przeprowadza się porównanie białka nienaruszonego, po denaturacji cieplnej oraz po strawieniu przez pepsynę pod względem wiązania IgE, ponieważ zmieniona strawność może mieć wpływ na alergenność nowego białka ulegającego ekspresji.
d) Badania dodatkowe
Mimo że dodatkowe badania, w tym oznaczenia in vitro wykorzystujące hodowle komórkowe lub testy in vivo na modelach zwierzęcych nie zostały dotychczas zatwierdzone do celów regulacyjnych, mogą one dostarczyć pożytecznych informacji dodatkowych, na przykład dotyczących możliwości wywoływania ponownych przypadków uwrażliwienia przez nowe białko ulegające ekspresji.
1.5.2. Ocena alergenności genetycznie zmodyfikowanej żywności lub paszy
Kiedy wiadomo, że roślina biorca jest alergenna, wnioskodawca ocenia wszelkie potencjalne zmiany alergenności genetycznie zmodyfikowanej żywności lub paszy poprzez porównanie jej zestawu alergenów z zestawem alergenów jej tradycyjnego odpowiednika. Należy w szczególności zbadać możliwość nadekspresji naturalnych, endogennych alergenów w roślinach zmodyfikowanych genetycznie.
Wnioskodawca stosuje podejście oparte na analizie poszczególnych przypadków w zależności od dostępnych informacji na temat potencjału alergennego rośliny biorcy. Zazwyczaj przeprowadza się to metodami analitycznymi, takimi jak proteomika w połączeniu z zastosowaniem jako próbników surowic pochodzących od ludzi cierpiących na alergie. Surowice pochodzące od osób cierpiących na alergie dobrze scharakteryzowane w warunkach klinicznych, które są materiałami odniesienia dla badań wiązania IgE, mogą być dostępne w ograniczonej liczbie i w niewielkich ilościach. Aby zminimalizować wykorzystanie surowic ludzkich, można uzyskać istotne informacje wstępne dotyczące prawdopodobieństwa niezamierzonej zmiany całkowitej alergenności rośliny zmodyfikowanej genetycznie dzięki wykorzystaniu surowic zwierzęcych uwrażliwionych eksperymentalnie w dobrze zdefiniowanych warunkach oraz włączeniu do analizy porównawczej składu odpowiednich zidentyfikowanych alergenów endogennych.
Wnioskodawca dostarcza ponadto informacji na temat rozpowszechnienia alergii u osób pracujących przy uprawie danej rośliny zmodyfikowanej genetycznie, mających z nią kontakt lub przebywających w jej sąsiedztwie, o ile takie dane są dostępne.
1.5.3. Adiuwancyjność
Adiuwanty to substancje, które podawane wraz z antygenem zwiększają reakcję immunologiczną na ten antygen, a zatem mogą wzmacniać również reakcję alergiczną. W przypadkach gdy znane aspekty funkcjonalne nowego białka ulegającego ekspresji lub podobieństwo strukturalne do znanych silnych adiuwantów może wskazywać na możliwe działanie adiuwancyjne, wnioskodawca ocenia potencjalną rolę adiuwancyjną tych białek. Podobnie jak w przypadku alergenów, interakcje z innymi składnikami materiału żywnościowego lub jego obróbka mogą zmienić strukturę i biodostępność danego adiuwantu i zmienić jego aktywność biologiczną.
1.5.4. Wniosek z oceny alergenności
W podsumowaniu oceny alergenności stwierdza się, czy:
a) nowe białka mogą być alergenne;
b) genetycznie zmodyfikowana żywność lub pasza mogą być bardziej alergenne niż ich tradycyjne odpowiedniki.
W przypadku gdy istnieje prawdopodobieństwo większej alergenności na skutek modyfikacji genetycznej, genetycznie zmodyfikowana żywność lub pasza podlegają dalszemu opisowi z uwzględnieniem przewidywanego spożycia (zob. sekcja 2). Wnioskodawca proponuje warunki wprowadzenia do obrotu (takie jak monitorowanie po wprowadzeniu do obrotu i etykietowanie).
1.6. Ocena wartości odżywczej
1.6.1. Cele oceny wartości odżywczej
Wnioskodawca dostarcza ocenę wartości odżywczej, aby wykazać, że:
a) wprowadzenie do obrotu genetycznie zmodyfikowanej żywności i paszy nie będzie niekorzystne pod względem wartości odżywczej ani dla ludzi, ani dla zwierząt. Ocena ta musi zawierać znaczenie dla wartości odżywczej pokarmu nowych białek ulegających ekspresji, innych nowych składników oraz zmian w poziomach składników żywności i paszy, jak również możliwych zmian w całej diecie konsumenta lub zwierzęcia;
b) niezamierzone skutki modyfikacji genetycznych, które zostały zidentyfikowane lub których wystąpienie można założyć na podstawie uprzednich analiz molekularnych, składu lub fenotypu zgodnie z sekcjami 1.2 i 1.3, nie wpłynęły ujemnie na wartość odżywczą genetycznie zmodyfikowanej żywności i paszy.
W przypadku złożonych modyfikacji genetycznych, które nastąpiły na skutek tradycyjnego krzyżowania, wnioskodawca dostarcza ocenę potencjalnych zmian wartości odżywczej, które mogły nastąpić na skutek synergicznego lub antagonistycznego działania produktów genowych, w tym zmian składu. Może to mieć szczególne znaczenie w przypadkach, w których połączona ekspresja nowo wprowadzonych genów ma nieoczekiwane skutki dla szlaków biochemicznych.
1.6.2. Punkty, które należy uwzględnić w ocenie wartości odżywczej genetycznie zmodyfikowanej żywności i paszy
Ocena wartości odżywczej genetycznie zmodyfikowanej żywności i paszy zawiera:
a) skład genetycznie zmodyfikowanej żywności i paszy przy uwzględnieniu zawartości składników odżywczych oraz substancji obniżających właściwości odżywcze pokarmów (zob. badania składu opisane w sekcji 1.3);
b) biodostępność i skuteczność biologiczną składników odżywczych żywności i paszy przy uwzględnieniu potencjalnego wpływu transportu, przechowywania oraz przewidywanego sposobu obróbki żywności i paszy;
c) przewidywane spożycie żywności i paszy w diecie (zob. sekcja 2) i wynikający z tego wpływ na odżywianie.
Jeżeli w wyniku analizy porównawczej zostaną zidentyfikowane cechy składu genetycznie zmodyfikowanej żywności i paszy inne niż u ich tradycyjnych odpowiedników lub nierównoważne cechom odmian wzorcowych, ich przydatność do celów żywieniowych ocenia się na podstawie aktualnej wiedzy naukowej. Jeżeli ocena ta doprowadzi do wniosku, że genetycznie zmodyfikowana żywność i pasza są pod względem wartości odżywczej równoważne z tradycyjnymi odpowiednikami, nie przeprowadza się dalszych badań. W przeciwnym przypadku, jeżeli na podstawie oceny informacji uzyskanych na drodze analizy porównawczej nie można wyciągnąć wniosków co do równoważności pod względem wartości odżywczej, należy przeprowadzić dalsze badania wartości odżywczej. Badania porównawcze wzrostu prowadzone są na młodych osobnikach szybko rosnących gatunków zwierząt (takich jak brojlery w przypadku zwierząt innych niż przeżuwacze; jagnięta w przypadku przeżuwaczy; lub inne szybko rosnące gatunki).
1.6.3. Badania wartości odżywczej genetycznie zmodyfikowanej żywności
Wnioskodawca określa potrzebę oraz program badania wartości odżywczej na podstawie rodzaju wprowadzonych cech, wyniku analizy porównawczej oraz, w stosownych przypadkach, 90-dniowych badań żywieniowych. Dodatkowe informacje dotyczące wartości odżywczej można uzyskać z badań porównawczych wydajności wzrostu prowadzonych na zwierzętach innych gatunków, takich jak brojlery, dotyczących oceny wartości odżywczej genetycznie zmodyfikowanej paszy. Dieta grupy kontrolnej podczas badania wartości odżywczej powinna zawierać tradycyjny odpowiednik oraz, tam gdzie to potrzebne, dodatkowe okazy porównawcze. W przypadku roślin zmodyfikowanych genetycznie tolerujących herbicydy badany materiał powinien pochodzić ze zmodyfikowanej genetycznie rośliny wystawionej na działanie przeznaczonego dla niej herbicydu.
Żywność genetycznie zmodyfikowana po to, aby dostarczyć konsumentowi dodatkowe korzyści zdrowotne w porównaniu z żywnością tradycyjną, może być korzystna tylko dla szczególnej populacji lub jej części, a dla innych jej spożycie może być ryzykowne. W przypadkach, w których należy ustalić zmienioną biodostępność mogącą stanowić ryzyko dla części populacji, oznacza się zawartość danego składnika odżywczego w żywności przy uwzględnieniu wszystkich form danego związku. Metodę badania biodostępności należy wybrać oddzielnie dla każdego przypadku, w zależności od danego składnika odżywczego lub innego składnika, żywności zawierającej te składniki, jak również stanu zdrowia, odżywienia i zwyczajów żywieniowych konkretnej populacji, która miałaby spożywać daną żywność.
1.6.4. Badania wartości odżywczej genetycznie zmodyfikowanej paszy
Wnioskodawca określa potrzebę oraz program dalszego badania wartości odżywczej na podstawie rodzaju wprowadzonych cech, wyniku analizy porównawczej oraz 90-dniowych badań żywieniowych, o ile są dostępne. Dodatkowe informacje dotyczące wartości odżywczej można uzyskać z badań porównawczych wydajności wzrostu prowadzonych na zwierzętach innych gatunków, takich jak brojlery, dotyczących oceny wartości odżywczej genetycznie zmodyfikowanej paszy. Dieta grupy kontrolnej podczas badania wartości odżywczej powinna zawierać tradycyjny odpowiednik oraz, tam gdzie to potrzebne, dodatkowe okazy porównawcze.
W przypadku genetycznie zmodyfikowanej paszy o poprawionych cechach odżywczych w celu oceny wpływu na żywienie przeprowadza się badanie żywieniowe na zwierzętach, od których lub z których pozyskuje się żywność, należących do gatunku, dla którego pasza jest przeznaczona. W przypadku roślin zmodyfikowanych genetycznie o zwiększonej zawartości i biodostępności składników odżywczych przeprowadza się badania na zwierzętach, od których lub z których pozyskuje się żywność, należących do gatunku, dla którego pasza jest przeznaczona, aby określić biodostępność poszczególnych składników odżywczych rośliny zmodyfikowanej genetycznie w porównaniu z jej tradycyjnym odpowiednikiem. W przypadku roślin zmodyfikowanych genetycznie o cechach zmienionych w celu poprawienia wydajności hodowli zwierząt poprzez zwiększenie zawartości składników odżywczych (np. zwiększenie zawartości oleju) lub zwiększenie zawartości konkretnego składnika (np. ważnego aminokwasu czy witaminy), należy opracować odpowiednią dietę kontrolną przy użyciu tradycyjnego odpowiednika i suplementacji danym składnikiem odżywczym w ilości odpowiadającej zmianie dokonanej w roślinie zmodyfikowanej genetycznie. Produkty uboczne (takie jak mączki z nasion oleistych), z których został wyekstrahowany składnik będący celem modyfikacji genetycznej, można porównać z produktami ubocznymi wyprodukowanymi z tradycyjnego odpowiednika.
Okres badań żywieniowych na zwierzętach, dla których pasza jest przeznaczona, obejmuje okres wzrostu lub okres tuczu końcowego przed ubojem w przypadku kurcząt, świń oraz bydła opasowego, lub większą część cyklu laktacji dla krów mlecznych, bądź cyklu składania jaj dla kur niosek lub przepiórek. W przypadku pasz przeznaczonych tylko dla akwakultury należy przeprowadzić badania wzrostu na gatunkach wodnych takich jak karp, zębacz, łososiowate lub inny gatunek typowo roślinożerny.
W stosownych przypadkach należy przeprowadzić badania według różnych schematów doświadczalnych, aby wykazać, że roślina zmodyfikowana genetycznie o poprawionych właściwościach odżywczych posiada oczekiwaną wartość odżywczą. Dokładny program doświadczeń oraz podejście statystyczne do doświadczeń karmienia zwierząt, od których lub z których pozyskuje się żywność, mające na celu zbadanie wartości odżywczej genetycznie zmodyfikowanej paszy o poprawionych właściwościach odżywczych zależy od gatunku zwierzęcia, dla którego pasza jest przeznaczona, rodzaju badanych cech rośliny oraz wielkości oczekiwanego efektu. Diety eksperymentalne opracowuje się w taki sposób, aby kluczowe mierzone punkty końcowe zależały od różnicy w ilości lub dostępności badanego składnika. Pomiary punktów końcowych powinny zależeć od gatunku używanego do badania, ale muszą obejmować spożycie paszy, masę ciała, wydajność hodowli zwierząt oraz biodostępność składników odżywczych.
Bardziej szczegółowe wytyczne co do stosowania wymogów niniejszej sekcji można znaleźć w sprawozdaniu działającej przy EFSA grupy roboczej panelu GMO ds. prób żywieniowych na zwierzętach (10).
1.6.5. Wniosek z oceny wartości odżywczej
We wniosku z oceny wartości odżywczej genetycznie zmodyfikowanej żywności i paszy stwierdza się, czy wartości odżywcze danej genetycznie zmodyfikowanej żywności i paszy i jej tradycyjnego odpowiednika są równoważne, przy uwzględnieniu naturalnej zmienności.
Wnioskodawca analizuje wynik oceny wartości odżywczej przy uwzględnieniu przewidywanego spożycia genetycznie zmodyfikowanej żywności i paszy (zob. sekcja 2).
1.7. Standardowe wytyczne dla badań toksyczności
Do badań toksyczności wnioskodawca stosuje uzgodnione na arenie międzynarodowej wytyczne i metody badań opisane w rozporządzeniu Komisji (WE) nr 440/2008 z dnia 30 maja 2008 r. ustalającym metody badań zgodnie z rozporządzeniem (WE) nr 1907/2006 Parlamentu Europejskiego i Rady w sprawie rejestracji, oceny, udzielania zezwoleń i stosowanych ograniczeń w zakresie chemikaliów (REACH) (11) (zob. tabele 1 i 2). W tabelach 1 i 2 znajduje się niewyczerpujący wykaz zatwierdzonych metod badań, które w razie potrzeby i po ewentualnej adaptacji należy stosować w badaniach toksykologicznych GMO.
Wydajność metod badawczych zależy od rodzaju genetycznie zmodyfikowanej żywności i paszy, rodzaju modyfikacji genetycznej oraz będących jej skutkiem zamierzonych i niezamierzonych zmian, przewidywanego zastosowania i narażenia/spożycia, oraz dostępnej wiedzy. Niektóre z tych testów zostały opracowane w celu oceny zagrożeń w miejscu pracy (zob. sekcje 1.4 i 1.5).
Tabela 1
Niewyczerpujący wykaz zatwierdzonych metod badawczych określonych w rozporządzeniu (WE) nr 440/2008, które po ewentualnej adaptacji należy stosować w badaniach toksykologicznych GMO
| Tytuł | Odniesienie do metody w części B załącznika do rozporządzenia (WE) nr 440/2008 |
| TOKSYCZNOŚĆ OSTRA (DERMALNA) | B.3. |
| SENSYBILIZACJA SKÓRY | B.6. |
| TOKSYCZNOŚĆ POWTARZANEJ (28 DNI) DAWKI (DOUSTNA) | B.7. |
| TOKSYCZNOŚĆ POWTARZANEJ (28 DNI) DAWKI (DERMALNA) | B.9. |
| BADANIE TOKSYCZNOŚCI PODPRZEWLEKŁEJ DROGĄ POKARMOWĄ – STUDIUM TOKSYCZNOŚCI NA GRYZONIACH METODĄ POWTARZANEJ 90-DNIOWEJ DAWKI DROGĄ POKARMOWĄ | B.26. |
| BADANIE TOKSYCZNOŚCI CHRONICZNEJ | B.30. |
| BADANIE RAKOTWÓRCZOŚCI | B32. |
| POŁĄCZONE BADANIE TOKSYCZNOŚCI CHRONICZNEJ/RAKOTWÓRCZOŚCI | B.33. |
| BADANIE TOKSYCZNOŚCI REPRODUKCJI JEDNEGO POKOLENIA | B.34. |
| DWUPOKOLENIOWE BADANIE TOKSYCZNOŚCI REPRODUKCYJNEJ | B.35. |
| TOKSYKOKINETYKA | B.36. |
| BADANIE NEUROTOKSYCZNOŚCI U GRYZONI | B.43. |
Tabela 2
Badania genotoksyczności określone w rozporządzeniu (WE) nr 440/2008
| Tytuł | Odniesienie do metody w części B załącznika do rozporządzenia (WE) nr 440/2008 |
| MUTAGENNOŚĆ – BADANIE ABERRACJI CHROMOSOMOWEJ SZPIKU KOSTNEGO IN VIVO | B.11. |
| MUTAGENNOŚĆ – BADANIE MIKROJĄDROWE ERYTROCYTÓW IN VIVO U SSAKÓW | B.12. |
| MUTAGENNOŚĆ: BADANIE MUTACJI POWROTNYCH W KOMÓRKACH BAKTERYJNYCH | B.13/14. |
| BADANIE MUTAGENNOŚCI I BADANIE PRZESIEWOWE NA RAKOTWÓRCZOŚĆ MUTACJA GENU – SACCHAROMYCES CEREVISIAE | B.15. |
| BADANIE REKOMBINACJI MITOTYCZNEJ – SACCHAROMYCES CEREVISIAE | B.16. |
| USZKODZENIE I NAPRAWA DNA – NIEREGULARNA SYNTEZA DNA – KOMÓRKI SSAKÓW IN VITRO | B.18. |
| MUTAGENNOŚĆ – BADANIE MUTACJI GENETYCZNEJ IN VITRO U SSAKÓW | B.17. |
| TEST WYMIANY CHROMATYD SIOSTRZANYCH IN VITRO | B.19. |
| BADANIE PRZEMIANY KOMÓRKOWEJ SSAKÓW IN VITRO | B.21. |
| BADANIE ABERRACJI CHROMOSOMOWEJ SPERMATOGONIÓW U SSAKÓW | B.23. |
2. OCENA NARAŻENIA – PRZEWIDYWANE SPOŻYCIE/ZAKRES ZASTOSOWANIA
Oszacowanie przewidywanego spożycia powinno być podstawowym elementem oceny ryzyka genetycznie zmodyfikowanej żywności i paszy i jest również wymagane dla oceny wartości odżywczej. Wnioskodawca dostarcza informacje dotyczące przewidywanej funkcji, roli w diecie oraz oczekiwanej skali stosowania genetycznie zmodyfikowanej żywności i paszy w UE. Ponadto dostarcza się przewidywany zakres stężeń nowo produkowanych białek lub istniejących białek roślinnych celowo zmodyfikowanych w genetycznie zmodyfikowanej żywności i paszy, które mają zostać wprowadzone do obrotu.
Na podstawie reprezentatywnych danych dotyczących konsumpcji produktów uzyskanych z roślin będących tradycyjnymi odpowiednikami wnioskodawca szacuje przewidywane średnie i maksymalne spożycie genetycznie zmodyfikowanej żywności i paszy. Można zastosować metody probabilistyczne do wyznaczenia zakresu prawdopodobnych wartości zamiast wartości pojedynczych czy oszacowań punktowych. Wnioskodawca musi zidentyfikować i wziąć pod uwagę szczególne grupy w populacji unijnej, które prawdopodobnie będą bardziej narażone, i uwzględnić to narażenie w ocenie ryzyka. Należy opisać wszelkie założenia poczynione w ocenie narażenia. Należy korzystać z najnowszych metodologii oraz odpowiednich danych dotyczących konsumpcji. Dane dotyczące importu i wielkości produkcji mogą zawierać dodatkowe informacje przydatne do oceny spożycia.
Wnioskodawca za pomocą odpowiednich metod określa stężenia nowych białek ulegających ekspresji, innych nowych składników oraz endogennych składników żywności i paszy, których poziom uległ zmianie na skutek modyfikacji genetycznej (na przykład z powodu zmiany szlaków metabolicznych) w tych częściach rośliny zmodyfikowanej genetycznie, które są przeznaczone na żywność lub paszę. Należy oszacować oczekiwane spożycie tych składników przy uwzględnieniu wpływu przetwarzania, przechowywania i przewidywanej obróbki danej żywności i paszy, na przykład potencjalną akumulację lub redukcję. W przypadkach gdy modyfikacja genetyczna spowodowała zmianę zawartości składnika naturalnego lub gdy nowy składnik występuje naturalnie w innych produktach żywnościowych i paszowych, należy ocenić przewidywaną zmianę całkowitego spożycia tego składnika przy uwzględnieniu zarówno realistycznych, jak i najbardziej pesymistycznych scenariuszy spożycia.
Wnioskodawca dostarcza informacje na temat znanego lub przewidywanego spożycia przez ludzi/zwierzęta analogicznej genetycznie zmodyfikowanej żywności i paszy oraz na temat innych dróg narażenia na odpowiednie składniki nowe i naturalne, w tym na temat ilości, częstotliwości i innych czynników wpływających na narażenie.
3. CHARAKTERYSTYKA RYZYKA
3.1. Wprowadzenie
Wnioskodawca opiera swoją charakterystykę ryzyka roślin zmodyfikowanych genetycznie oraz genetycznie zmodyfikowanej żywności i paszy na danych uzyskanych z identyfikacji zagrożeń i charakterystyki zagrożeń oraz na danych dotyczących narażenia/spożycia. Przez uwzględnienie wszystkich dostępnych dowodów pochodzących z wielu analiz, w tym analizy molekularnej, fenotypowej, agronomicznej i analizy składu oraz badań toksyczności i alergenności, wnioskodawca dopilnowuje, aby charakterystyka ryzyka miała kompleksowy charakter. Wnioskodawca uwzględnia wskazania wynikające z charakterystyki ryzyka, które mogą wymagać określonych działań w zakresie monitorowania genetycznie zmodyfikowanej żywności i paszy po wprowadzeniu do obrotu.
Sporządzając charakterystykę ryzyka, wnioskodawca wykazuje, że identyfikacja zagrożeń i charakterystyka zagrożeń są kompletne. Wnioskodawca omawia jakość istniejących danych i informacji. W omówieniu wyraźnie wskazuje sposób, w jaki to źródło informacji zostało uwzględnione przy ustalaniu ostatecznej charakterystyki ryzyka.
Wnioskodawca przedstawia szacunki dotyczące niepewności związanej z każdym badaniem, jak również z różnymi etapami oceny ryzyka. Wnioskodawca w miarę możliwości ujmuje je ilościowo. Dokonuje się rozróżnienia pomiędzy niepewnością, która odzwierciedla naturalną zmienność parametrów biologicznych (w tym zmienność podatności w populacjach), a zmiennością reakcji u różnych gatunków.
W zależności od badanego zagadnienia i od dostępnych danych wnioskodawca sporządza jakościową oraz, w miarę możliwości, ilościową charakterystykę ryzyka. Warunki dotyczące szacowanego ryzyka i związanej z nim niepewności są przedstawiane możliwie precyzyjnie.
3.2. Kwestie brane pod uwagę przy charakterystyce ryzyka
W stosownych przypadkach i w zależności od rodzaju modyfikacji genetycznej wnioskodawca dokonuje oceny ryzyka w odniesieniu do roślin zmodyfikowanych genetycznie w sposób zintegrowany zgodnie z sekcją 3.1. Taką ocenę ryzyka przeprowadza się oddzielnie dla każdego przypadku, w zależności od zmodyfikowanej rośliny i rodzaju modyfikacji genetycznej, metod prowadzenia upraw rośliny zmodyfikowanej genetycznie i zastosowań genetycznie zmodyfikowanej żywności i paszy. Wnioskodawca bierze pod uwagę różne kwestie rozważane w odniesieniu do identyfikacji zagrożeń, charakterystyki zagrożeń i etapów narażenia. Wyniki rozważania tych kwestii wnioskodawca uwzględnia łącznie na etapie charakterystyki ryzyka. Wykaz kwestii podany w niniejszej sekcji nie jest wyczerpujący.
3.2.1. Charakterystyka molekularna
Ocena właściwości i dotychczasowych zastosowań rośliny dawcy i biorcy jest najważniejszym elementem przy określaniu potrzeby przeprowadzenia konkretnych analiz, takich jak analiza występowania określonych toksyn lub alergenów w niezmodyfikowanej roślinie biorcy, których poziom może być zwiększony w sposób niezamierzony w wyniku modyfikacji genetycznej.
Wnioskodawca omawia protokoły transformacji, strategie charakterystyki molekularnej oraz specyficzność i czułość wykorzystanych metod w odniesieniu do celowej lub niezamierzonej insercji oraz ekspresji sekwencji genów.
Jeżeli w drodze analizy sekwencji określono potencjalne zagrożenie, wnioskodawca wykazuje sposób, w jaki rodzaje podejścia, takie jak analiza bioinformatyczna, analiza składu/agronomiczna i ewentualne badania żywieniowe na zwierzętach, przeprowadzone w odniesieniu do całej genetycznie zmodyfikowanej żywności i paszy, przyczyniają się do oceny bezpieczeństwa. Wartość uzyskanych wyników ocenia się w świetle dostępnej wiedzy na temat struktury i funkcji genomowych baz danych przedmiotowych gatunków upraw lub gatunków spokrewnionych.
W przypadku genetycznie zmodyfikowanych roślin zawierających złożone modyfikacje genetyczne ocenia się dodatkowe ryzyko mogące wynikać z połączonych skutków kombinacji genów.
3.2.2. Analiza porównawcza
Pierwszym celem analizy porównawczej jest określenie ewentualnych różnic między rośliną zmodyfikowaną genetycznie a jej tradycyjnym odpowiednikiem oraz, w stosownych przypadkach, dodatkowymi okazami porównawczymi. Drugim celem analizy porównawczej jest ustalenie ewentualnego braku równoważności między rośliną zmodyfikowaną genetycznie a jej odmianami wzorcowymi. Te różnice lub brak równoważności należy oceniać pod względem ich ewentualnego wpływu na bezpieczeństwo i właściwości odżywcze żywności i paszy, biorąc pod uwagę naturalną zmienność. Szacowane ryzyko i związaną z nim niepewność należy ustalać możliwie najdokładniej i brać je pod uwagę.
Wnioskodawca wykazuje, że analiza porównawcza rośliny zmodyfikowanej genetycznie i jej tradycyjnego odpowiednika w odniesieniu do cech agronomicznych, morfologicznych i składu została przeprowadzona zgodnie z wymogami niniejszego rozporządzenia. Uzasadnia się wybór tradycyjnego odpowiednika i, w stosownych przypadkach, dodatkowych okazów porównawczych.
3.2.3. Bezpieczeństwo żywności i paszy w odniesieniu do spożycia
Wnioskodawca ocenia wygenerowane dane, aby oszacować ewentualne krótkotrwałe i długofalowe ryzyko dla zdrowia ludzi lub zwierząt związane ze spożywaniem genetycznie zmodyfikowanej żywności lub paszy w odniesieniu do ekspresji nowych białek/metabolitów, a także znacznie zmienione poziomy pierwotnych białek/metabolitów rośliny w genetycznie zmodyfikowanej żywności/paszy. Ocena ta obejmuje dogłębną analizę znaczenia i ograniczeń każdego z badań, a także całości informacji.
Wnioskodawca rozpatruje zakres poziomów obserwowanych dla związków, co do których wiadomo, że występują w tradycyjnym odpowiedniku i w odmianach wzorcowych. Ta zmienność może być spowodowana różnicami, które są zależne od genotypu, zależne od środowiska lub być wywołana wzajemnym oddziaływaniem genotypu i środowiska. Ponadto można uwzględnić zakres poziomów obserwowanych w szerokim spektrum żywności i paszy reprezentatywnych dla diety ludzi i zwierząt, zważywszy że odzwierciedla poziomy określonego związku, na które konsumenci mogą być narażeni.
Jeżeli przy pomocy określonych badań wykaże się, że pojedynczy składnik lub cała genetycznie zmodyfikowana żywność lub pasza wywołują szkodliwy skutek, przedstawia się informacje na temat zależności dawka-odpowiedź, wartości progowych, opóźnionego wystąpienia szkodliwych skutków, ryzyka dla określonych grup w populacji, wykorzystania czynników niepewności do ekstrapolacji danych z badań na zwierzętach na ludzi.
Wnioskodawca uwzględnia dane dotyczące właściwości nowych związków obecnych w roślinie zmodyfikowanej genetycznie, w tym potencjalne skutki biologiczne dla ludzi i zwierząt. Jeśli dane związki mają znane szkodliwe skutki związku dla zdrowia, a w przepisach szczegółowych przewidziane są najwyższe dopuszczalne poziomy obecności tych związków w roślinie lub jej produktach, bierze się pod uwagę te najwyższe dopuszczalne poziomy. W przeciwnym razie w odniesieniu do przewidywanego spożycia uwzględnia się wartości referencyjne dopuszczalnych lub tolerowanych poziomów spożycia, takie jak dopuszczalne dzienne spożycie lub górny tolerowany poziom spożycia. W przypadku gdy związek był bezpiecznie spożywany w żywności, poziomy spożycia przez konsumentów w konwencjonalnej diecie uważa się za bezpieczne.
Wnioskodawca ocenia informacje dotyczące skutków przetwarzania dla nowych związków. Uwzględnia się potencjalną akumulację/zmniejszenie ilości danego związku w żywności i paszy wchodzących w skład diety ludzi lub zwierząt. Wnioskodawca ocenia również znaczenie różnic wynikających z reakcji chemicznych, o których wiadomo, że zachodzą w warunkach przetwarzania.
W przypadku bardziej skomplikowanych modyfikacji genetycznych, np. transferu wielu genów w jednym konstrukcie, retransformacji wcześniej istniejących genetycznie zmodyfikowanych linii oraz łączenia modyfikacji genetycznych poprzez tradycyjne metody rozmnażania genetycznie zmodyfikowanych roślin rodzicielskich, wnioskodawca omawia strategie oceny wszelkiego ryzyka związanego z możliwym wzajemnym oddziaływaniem pomiędzy nowymi białkami ulegającymi ekspresji, nowymi metabolitami i oryginalnymi składnikami rośliny. W ocenie uwzględnia się wszystkie dostępne informacje, w tym na temat sposobu działania nowych białek ulegających ekspresji, właściwości molekularnych i cech składu/agronomicznych rośliny zmodyfikowanej genetycznie oraz wyników badań toksyczności i żywieniowych na zwierzętach.
Wnioskodawca dokonuje oceny uzyskanych danych w celu oszacowania potencjalnej alergenności nowych białek ulegających ekspresji w roślinach zmodyfikowanych genetycznie w odniesieniu do wprowadzenia nowych białek alergennych do roślin wykorzystywanych w żywności i paszy, które to białka mogą wywołać reakcje alergiczne u osób wrażliwych, a także ocenia informacje w celu wykazania, że modyfikacja genetyczna nie wywołuje niepożądanych zmian we właściwościach lub poziomach ekspresji endogennych białek alergennych w genetycznie zmodyfikowanej żywności. Uzasadnia się w szczególności wybór modeli testowych pod względem swoistości, przewidywalności i statusu walidacji.
W odniesieniu do szacowanego spożycia genetycznie zmodyfikowanej żywności wnioskodawca ocenia zastosowaną metodykę pod względem niepewności związanej z prognozami dotyczącymi długotrwałego spożywania. Szczególną uwagę zwraca się na te rośliny zmodyfikowane genetycznie, których celem jest zmiana właściwości odżywczych żywności i paszy. W przypadku tych produktów zmodyfikowanych genetycznie wymóg monitorowania produktów po wprowadzeniu do obrotu jest omawiany jako mechanizm ustalania faktycznych zmian w ogólnych dietetycznych wzorcach spożywania genetycznie zmodyfikowanej żywności, zakresu tych zmian oraz tego, czy produkt powoduje wystąpienie znanych skutków (ubocznych) lub nieoczekiwanych skutków ubocznych. Jeżeli prowadzenie monitorowania produktu po wprowadzeniu do obrotu uznaje się za konieczne, podaje się informacje na temat wiarygodności, czułości i swoistości proponowanych metod.
3.3. Wyniki charakterystyki ryzyka
Zgodnie z wymogami art. 4 i 16 rozporządzenia (WE) nr 1829/2003 wnioskodawca dopilnowuje, aby ostateczna charakterystyka ryzyka wyraźnie wykazywała, że:
a) genetycznie zmodyfikowana żywność i pasza nie ma szkodliwego wpływu na zdrowie ludzi i zwierząt;
b) genetycznie zmodyfikowana żywność nie różni się od żywności, którą ma zastępować, w takim zakresie, że jej normalne spożycie nie powoduje niekorzystnych skutków odżywczych dla konsumentów;
c) genetycznie zmodyfikowana żywność nie wprowadza konsumenta w błąd;
d) genetycznie zmodyfikowana pasza nie szkodzi konsumentowi ani nie wprowadza go w błąd z powodu pogorszenia szczególnych cech produktów zwierzęcych;
e) genetycznie zmodyfikowana pasza nie odbiega od paszy, którą ma zastąpić, w takim stopniu, że jej normalne spożycie nie powoduje szkodliwych skutków odżywczych dla zwierząt lub ludzi.
Wnioskodawca wyraźnie wskazuje założenia, jakie poczyniono w trakcie oceny ryzyka w celu przewidzenia prawdopodobieństwa wystąpienia i dotkliwości szkodliwych skutków w danej populacji oraz charakter i nasilenie niepewności związanej z określeniem tego ryzyka.
Wnioskodawca uwzględnia również szczegółowe informacje uzasadniające włączenie do wniosku propozycji etykietowania lub niezamieszczenie takiej propozycji, zgodnie z art. 13 ust. 2 lit. a) i art. 13 ust. 3 oraz art. 25 ust. 2 lit. c) i art. 25 ust. 3 rozporządzenia (WE) nr 1829/2003.
(1) Dz.U. L 43 z 14.2.1997, s. 1.
(2) Dziennik EFSA 2011; 9(5):2149.
(3) Dziennik EFSA 2010; 8(1):1250.
(4) Dziennik EFSA 2010; 8(1):1250.
(5) http://www.oecd.org/document/15/0,3746,en_2649_34385_ 46726799_1_1_1_1,00.html.
(6) http://www.efsa.europa.eu/en/efsajournal/pub/2760.htm.
(7) Dz.U. L 133 z 22.5.2008, s. 1.
(8) Dziennik EFSA 2011; 9(12):2438.
(9) Dziennik EFSA 2010; 8(7):1700.
(10) EFSA, 2008 Report of the EFSA GMO Panel Working Group on Animal Feeding Trials, 2008. Safety and nutritional assessment of GM plants and derived food and feed. The role of animal feeding trials. Food and Chemical Toxicology 46 (2008) S2–S70.
(11) Dz.U. L 142 z 31.5.2008, s. 1.
ZAŁĄCZNIK III
WALIDACJA METOD WYKRYWANIA, IDENTYFIKACJI I KWANTYFIKACJI MODYFIKACJI GENETYCZNEJ ORAZ WYMAGANIA DOTYCZĄCE PRÓBEK KONTROLNYCH I CERTYFIKOWANEGO MATERIAŁU ODNIESIENIA
1. WPROWADZENIE
1. Do celów wykonania art. 5 ust. 3 lit. i) i j) oraz art. 17 ust. 3 lit. i) i j) rozporządzenia (WE) nr 1829/2003 w niniejszym załączniku określono wymagania dotyczące:
a) charakterystyki wydajności przedstawionych metod;
b) wymogów technicznych co do rodzaju informacji, które wnioskodawca musi przedstawić, aby można było sprawdzić, czy te wymogi są spełnione;
c) próbek żywności i paszy oraz ich próbek kontrolnych;
d) certyfikowanego materiału odniesienia.
2. Wnioskodawca musi przedstawić informacje na temat metody jako takiej oraz testowania metody przeprowadzanego przez wnioskodawcę.
3. Wnioskodawca uwzględnia również dalsze wytyczne i informacje na temat procedur operacyjnych procesu walidacji udostępniane przez laboratorium referencyjne UE (EURL), o którym mowa w art. 32 rozporządzenia (WE) nr 1829/2003, wspomagane przez Europejską Sieć Laboratoriów GMO (1).
2. DEFINICJE
Do celów niniejszego załącznika stosuje się następujące definicje:
a) „certyfikowany materiał odniesienia” oznacza materiał odniesienia, o którym mowa w art. 5 ust. 3 lit. j) oraz w art. 17 ust. 3 lit. j) rozporządzenia (WE) nr 1829/2003, i odpowiada materiałowi lub substancji, których co najmniej jedna wartość właściwości jest certyfikowana na potrzeby kalibracji lub kontroli jakości metod. Towarzyszy mu certyfikat, w którym określona jest wartość konkretnej właściwości, związana z nią niepewność i podane jest oświadczenie o metrologicznej zgodności;
b) „ wymogi dotyczące wydajności metody” oznaczają minimalne kryteria wydajności, które zgodnie z przyjętymi na szczeblu międzynarodowym przepisami technicznymi metoda spełnia po przeprowadzeniu przez EURL badania walidacji.
3. WALIDACJA METODY
3.1. Informacje dotyczące metody
A. Zgodnie z art. 5 ust. 3 lit. i) oraz art. 17 ust. 3 lit. i) rozporządzenia (WE) nr 1829/2003 metoda odnosi się do wszystkich etapów metodycznych niezbędnych do przeprowadzenia analizy odnośnej żywności i paszy.
W odniesieniu do danej żywności lub paszy etapy metodyczne obejmują metody ekstrakcji DNA i późniejszej kwantyfikacji w układzie łańcuchowej reakcji polimerazy (PCR) w czasie rzeczywistym. W takim przypadku metodę stanowi cały proces od ekstrakcji do techniki PCR. Wnioskodawca przedstawia informacje na temat całej metody.
B. Wnioskodawca może odwoływać się do zwalidowanych protokołów, jeśli są dostępne i właściwe, na potrzeby modułów metod stosowanych w procedurze analitycznej, takich jak protokół ekstrakcji DNA z określonej matrycy.
W takim przypadku wnioskodawca przedstawia dane doświadczalne z własnej walidacji, w której w kontekście wniosku o zatwierdzenie pomyślnie zastosowano moduł metody.
C. Wnioskodawca wykazuje, że metoda spełnia następujące wymogi:
1. Metoda jest swoista dla modyfikacji genetycznej (dalej nazywana „ specyficzną dla danej modyfikacji”), a zatem działa wyłącznie w odniesieniu do organizmu zmodyfikowanego genetycznie lub produktu opartego na modyfikacji genetycznej i nie działa w przypadku jej zastosowania do innych modyfikacji genetycznych już zatwierdzonych; w przeciwnym przypadku metody nie można stosować na potrzeby jednoznacznego wykrywania/identyfikacji/kwantyfikacji. Powyższe wykazuje się w oparciu o dobór niecelowych, zatwierdzonych transgenicznych modyfikacji genetycznych i ich tradycyjnych odpowiedników. Badania te obejmują ściśle powiązane modyfikacje genetyczne.
2. Metodę stosuje się do próbek żywności lub paszy, do próbek kontrolnych oraz do certyfikowanego materiału odniesienia.
3. Do celów opracowania metody wykrywania wnioskodawca bierze pod uwagę następujące dokumenty:
a) Artykuły żywnościowe – Metody wykrywania organizmów zmodyfikowanych genetycznie i produktów pochodnych – Wymagania ogólne i definicje: ISO 24276;
b) Artykuły żywnościowe – Metody wykrywania organizmów zmodyfikowanych genetycznie i produktów pochodnych – Ekstrakcja kwasów nukleinowych: ISO 21571;
c) Artykuły żywnościowe – Metody wykrywania organizmów zmodyfikowanych genetycznie i produktów pochodnych – Metody ilościowe oparte na kwasach nukleinowych: ISO 21570;
d) Artykuły żywnościowe – Metody wykrywania organizmów zmodyfikowanych genetycznie i produktów pochodnych – Metody jakościowe oparte na kwasach nukleinowych: projekt normy europejskiej ISO 21569.
4. W metodzie uwzględnia się również bardziej szczegółowe wymogi określone we wspólnych kryteriach ustalonych przez EURL i ENGL w zakresie minimalnych wymogów dotyczących wydajności metod analitycznych w odniesieniu do badania GMO. Kryteria te stanowią część wytycznych zapewnionych przez EURL.
D. Do celów wykonania art. 5 ust. 3 lit. i) oraz art. 17 ust. 3 lit. i) rozporządzenia (WE) nr 1829/2003 wnioskodawca dostarcza ilościową metodę wykrywania materiału genetycznie zmodyfikowanego, specyficzną dla danej modyfikacji. Wnioskodawca przedstawia omówienie zasadności i ograniczeń dotyczących metod wykrywania w różnych typach żywności i paszy (różne matryce), które mają zostać wprowadzone do obrotu.
E. Wnioskodawca przedstawia pełny i szczegółowy opis metody.
Wnioskodawca w sposób niebudzący wątpliwości odnosi się do następujących zagadnień:
1) podstawa naukowa: wnioskodawca przedstawia w ogólnym zarysie zasady działania danej metody. Obejmuje to odniesienia do właściwych publikacji naukowych;
2) zakres metody: wnioskodawca wskazuje matrycę (np. przetworzona żywność, surowce), rodzaj próbek oraz zakres procentowy, w którym można stosować metodę;
3) właściwości robocze metody: określa się urządzenia wymagane do zastosowania metody w odniesieniu do analizy jako takiej oraz do przygotowania próbki. Podaje się także dalsze informacje dotyczące wszelkich szczególnych aspektów o podstawowym znaczeniu dla stosowania metody;
4) protokół: wnioskodawca przedstawia pełny, zoptymalizowany protokół dotyczący metody. Protokół zawiera wszystkie szczegóły wymagane do przeniesienia i zastosowania metody w sposób niezależny w innych laboratoriach;
5) podaje się szczegółowy opis modelu prognostycznego (lub podobnego narzędzia) niezbędnego do zinterpretowania wyników i wyciągnięcia wniosków. Wnioskodawca przedstawia instrukcje dotyczące właściwego zastosowania modelu;
6) wnioskodawca przedstawia programy rozmnażania, które mają być stosowane do produkcji genetycznie zmodyfikowanej żywności i paszy, oraz ich wpływ na interpretację wyników.
3.2. Informacje dotyczące testowania metody przeprowadzanego przez wnioskodawcę
A. Wnioskodawca przedstawia wszystkie dostępne i właściwe dane dotyczące optymalizacji oraz przeprowadzonego testowania metody. Jeśli jest to właściwe i możliwe, dane te i wyniki przedstawia się z wykorzystaniem parametrów wydajności, o których mowa w pkt 3.1.C.4. Wnioskodawca przedstawia również opis przeprowadzonego testowania i podstawowych wyników, a także dane obejmujące wartości odstające.
B. Wnioskodawca dopilnowuje, aby przedstawione informacje wykazywały odporność metody do celów wymiany między laboratoriami. W tym celu wnioskodawca przedstawia wyniki testowania metody przez co najmniej jedno laboratorium, inne niż to, które opracowało metodę.
C. Wnioskodawca przedstawia następujące informacje dotyczące opracowywania i optymalizacji metody:
1) przetestowane pary primerów i sondę, w stosownych przypadkach, w tym uzasadnienie, w jaki sposób i dlaczego wybrano sugerowaną parę primerów;
2) testowanie stabilności, które ustala się poprzez przedstawienie wyników doświadczalnych z testowania metody za pomocą innych odmian roślin;
3) swoistość, którą ustala się poprzez przedłożenie pełnej sekwencji insertu, w standardowym formacie elektronicznym, wraz z bazowymi parami sekwencji flankujących rośliny gospodarza, tak by umożliwić EURL dokonanie oceny swoistości proponowanej metody przez przeprowadzenie badania homologiczności w bazie danych molekularnych;
4) precyzja, przy czym względne odchylenie standardowe powtarzalności jest mniejsze niż lub równe 25 % masy w całym zakresie oznaczania metody.
D. Oprócz informacji wymaganych na podstawie sekcji A, B i C wnioskodawca przedstawia następujące informacje na temat testowania:
1) laboratoria uczestniczące, czas analizy oraz schemat doświadczenia, w tym szczegóły dotyczące liczby prób, próbek, replik itp.;
2) opis próbek laboratoryjnych (np. wielkość, jakość, datę pobrania), kontrole pozytywne i negatywne, a także certyfikowany materiał odniesienia, zastosowane plazmidy i tym podobne;
3) opis podejścia zastosowanego do analizy wyników i wartości odstających testu;
4) wszelkie obserwacje poczynione podczas testowania;
5) odniesienia do właściwej literatury lub przepisów technicznych wykorzystywanych podczas testowania.
3.3. Próbki żywności i paszy oraz ich próbki kontrolne
Do celów wykonania art. 5 ust. 3 lit. j) oraz art. 17 ust. 3 lit. j) rozporządzenia (WE) nr 1829/2003 wnioskodawca wraz z informacjami wymaganymi na podstawie sekcji 1, 2 i 3 niniejszego załącznika przedstawia również próbki żywności i paszy, a także ich próbki kontrolne z rodzaju oraz w ilości określanej przez EURL, a dotyczące danego wniosku o zatwierdzenie.
W informacjach dołączonych do próbek kontrolnych znajdują się informacje na temat hodowli roślin wykorzystanych do produkcji próbek kontrolnych oraz zygotyczności insertów.
Wnioskodawca może korzystać z tego samego surowca do produkcji certyfikowanego materiału odniesienia i do produkcji próbek kontrolnych.
4. CERTYFIKOWANY MATERIAŁ ODNIESIENIA
Certyfikowany materiał odniesienia wytwarza się zgodnie z Wytycznymi ISO 34 (Ogólne wymogi dotyczące kompetencji producentów materiału odniesienia) przez producenta akredytowanego zgodnie z Wytycznymi ISO 34.
Wnioskodawca przedstawia informacje na temat miejsca, w którym dostępny jest certyfikowany materiał odniesienia. Informacjom tym towarzyszą odpowiednie informacje wykazujące, że dostępność certyfikowanego materiału odniesienia zostanie utrzymana przez cały okres ważności zezwolenia. Do celów weryfikacji i oceny wartości stosuje się metodę zwalidowaną we właściwy sposób (zob. ISO/IEC 17025: Ogólne wymagania dotyczące kompetencji laboratoriów badawczych i wzorcujących).
Niepewność ocenia się zgodnie z Wytycznymi ISO dotyczącymi wyrażania niepewności pomiaru (GUM).
Główne właściwości tych przepisów technicznych przyjętych na szczeblu międzynarodowym są następujące:
1. Pojemniki na genetycznie zmodyfikowany materiał odniesienia:
a) pojemniki na genetycznie zmodyfikowany materiał odniesienia (takie jak butelki, fiolki, ampułki) są szczelne i zawierają nie mniej niż ustaloną ilość materiału;
b) zapewniona jest zamienność genetycznie zmodyfikowanego materiału odniesienia;
c) opakowanie jest odpowiednie do przeznaczenia;
d) etykieta jest właściwa i dobrej jakości.
2. Testowanie jednorodności:
a) próbki wykazują właściwą jednorodność;
b) badana jest jednorodność między butelkami;
c) w ogólnej, szacowanej niepewności dotyczącej materiału odniesienia uwzględnia się każdą możliwą niejednorodność stwierdzoną między butelkami. Wymóg ten ma zastosowanie nawet wtedy, gdy między butelkami nie występuje zmienność istotna statystycznie. W takim przypadku zmienność metody lub rzeczywista wyliczona zmienność między butelkami (w zależności od tego, która z nich jest większa) włącza się w ogólną niepewność.
3. Testowanie stabilności:
a) próbki wykazują właściwą stabilność;
b) stabilność wykazuje się za pomocą właściwej ekstrapolacji statystycznej dotyczącej okresu trwałości genetycznie zmodyfikowanego materiału odniesienia, mieszczącej się w granicach ustalonej niepewności; niepewność ta stanowi zazwyczaj część szacowanej niepewności dotyczącej materiału odniesienia. Przypisane wartości obowiązują wyłącznie w ograniczonym terminie i poddawane są monitorowaniu pod względem stabilności.
4. Charakterystyka partii:
1. Metody zastosowane w celu weryfikacji i certyfikacji:
a) są stosowane w warunkach ważnych metrologicznie;
b) zostały zwalidowane przed zastosowaniem w sposób technicznie właściwy;
c) są stosowane w oparciu o stopień dokładności i poprawności zgodny z docelowym progiem niepewności.
2. Każdy zestaw pomiarów:
a) jest identyfikowalny względem ustalonych odniesień;
b) jeśli jest to możliwe, towarzyszy mu oświadczenie o stopniu niepewności.
3. Laboratoria uczestniczące:
a) posiadają kompetencje wymagane do wykonania zadania;
b) są w stanie uzyskać identyfikowalność względem wymaganych, ustalonych odniesień;
c) są w stanie oszacować niepewność pomiaru;
d) posiadają odpowiedni i właściwy system zapewniania jakości.
5. Przechowywanie końcowe:
1. Aby uniknąć degradacji po wyprodukowaniu próbki, przed rozpoczęciem pomiarów wszystkie próbki przechowuje się w warunkach przeznaczonych do przechowywania końcowego genetycznie zmodyfikowanego certyfikowanego materiału odniesienia.
2. W przeciwnym razie przewozi się je doraźnie, utrzymując w takich warunkach przechowywania, dla których wykazano, iż nie mają one wpływu na przypisane wartości.
6. Ustanowienie certyfikatu dla certyfikowanego materiału odniesienia:
Ustanawia się certyfikat uzupełniony sprawozdaniem z certyfikacji, zawierający wszelkie istotne i wymagane przez użytkownika informacje.
Certyfikat i sprawozdanie są udostępniane, gdy genetycznie zmodyfikowany certyfikowany materiał odniesienia jest rozprowadzany.
W informacjach dołączonych do certyfikowanego materiału odniesienia znajdują się informacje na temat hodowli roślin wykorzystanych do produkcji certyfikowanego materiału odniesienia oraz zygotyczności insertów.
Wartość certyfikowana zawartości GMO podana jest jako ułamek masy oraz, jeżeli jest ona dostępna, w liczbie kopii na ekwiwalent genomu haploidalnego.
Certyfikowane wartości (takie jak ilość materiału genetycznie zmodyfikowanego wyrażona ułamkiem masy) są identyfikowalne względem ustalonych odniesień i towarzyszy im oświadczenie o niepewności rozszerzonej ważne dla całego okresu trwałości genetycznie zmodyfikowanego certyfikowanego materiału odniesienia.
(1) http://gmo-crl.jrc.ec.europa.eu/guidancedocs.htm.
- Data ogłoszenia: 2013-06-08
- Data wejścia w życie: 2013-06-28
- Data obowiązywania: 2013-06-28

- ROZPORZĄDZENIE KOMISJI (WE) NR 1981/2006 z dnia 22 grudnia 2006 r. ustalające szczegółowe zasady wykonania przepisów art. 32 rozporządzenia (WE) nr 1829/2003 Parlamentu Europejskiego i Rady w odniesieniu do wspólnotowego laboratorium referencyjnego dla organizmów zmodyfikowanych genetycznie
- ROZPORZĄDZENIE KOMISJI (WE) NR 641/2004 z dnia 6 kwietnia 2004 r. w sprawie szczegółowych zasad wykonywania rozporządzenia (WE) nr 1829/2003 Parlamentu Europejskiego i Rady odnoszącego się do wniosków o zatwierdzenie nowego typu żywności i paszy genetycznie zmodyfikowanej, powiadamiania o istniejących produktach, oraz przypadkowym lub technicznie nieuniknionym występowaniu materiału genetycznie zmodyfikowanego, który pomyślnie przeszedł ocenę ryzyka
REKLAMA
Akty ujednolicone
REKLAMA
REKLAMA