REKLAMA
Dziennik Ustaw - rok 2014 poz. 806
ROZPORZĄDZENIE
MINISTRA ZDROWIA1)
z dnia 3 czerwca 2014 r.
w sprawie sposobu i trybu sprawowania nadzoru nad bezpieczeństwem stosowania produktów leczniczych weterynaryjnych
Na podstawie art. 24 ust. 13 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne (Dz. U. z 2008 r. Nr 45, poz. 271, z późn. zm.2)) zarządza się, co następuje:
1) szczegółowy sposób i tryb sprawowania nadzoru nad bezpieczeństwem stosowania produktów leczniczych weterynaryjnych;
2) zakres danych objętych określonymi dokumentami, innymi niż formularz, o którym mowa w pkt 3, sporządzanymi w procesie sprawowania nadzoru nad bezpieczeństwem stosowania produktów leczniczych weterynaryjnych;
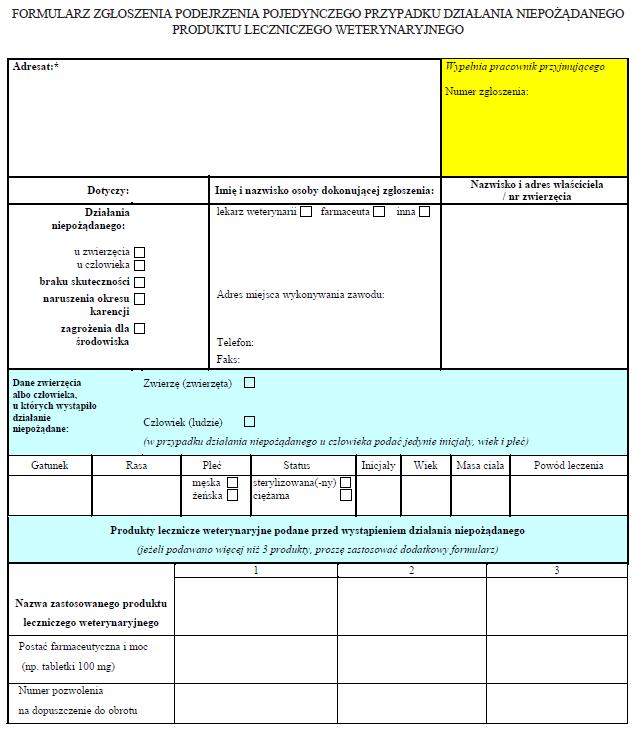
3) wzór formularza zgłoszenia pojedynczego przypadku działania niepożądanego produktu leczniczego weterynaryjnego.
1) zbiera i analizuje, w tym przeprowadza ocenę przyczynowo-skutkową, zgłoszenia pojedynczego przypadku działania niepożądanego produktu leczniczego weterynaryjnego, zwanego dalej „zgłoszeniem”, oraz raporty, o których mowa w art. 24 ust. 1 pkt 4 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, zwanej dalej „ustawą”, dotyczące:
a) pojedynczych przypadków działania niepożądanego produktu leczniczego weterynaryjnego po zastosowaniu produktu zgodnie z Charakterystyką Produktu Leczniczego Weterynaryjnego,
b) pojedynczych przypadków działania niepożądanego produktu leczniczego weterynaryjnego po zastosowaniu produktu niezgodnie z Charakterystyką Produktu Leczniczego Weterynaryjnego,
c) spodziewanego działania niepożądanego produktu leczniczego weterynaryjnego oraz zwiększonej częstotliwości jego występowania,
d) działania niepożądanego produktu leczniczego weterynaryjnego obserwowanego u ludzi, które wystąpiło w wyniku ekspozycji na ten produkt,
e) braku spodziewanej skuteczności produktu leczniczego weterynaryjnego,
f) przeniesienia, za pośrednictwem produktu leczniczego weterynaryjnego, czynnika zakaźnego,
g) zagrożeń dla środowiska związanych z zastosowaniem produktu leczniczego weterynaryjnego,
h) informacji, że wyznaczony okres karencji może być niewystarczający;
2) przekazuje niezwłocznie podmiotowi odpowiedzialnemu, w postaci elektronicznej, za pośrednictwem Eudravigilance Veterinary, o którym mowa w art. 51 akapit trzeci rozporządzenia (WE) nr 726/2004 Parlamentu Europejskiego i Rady z dnia 31 marca 2004 r. ustanawiającego wspólnotowe procedury wydawania pozwoleń dla produktów leczniczych stosowanych u ludzi i do celów weterynaryjnych i nadzoru nad nimi oraz ustanawiającego Europejską Agencję Leków (Dz. Urz. UE L 136 z 30.04.2004, str. 1; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 34, str. 229), zgłoszenia pochodzące z terytorium Rzeczypospolitej Polskiej, niepochodzące od podmiotu odpowiedzialnego, przy czym zgłoszenia dotyczące ciężkich niepożądanych działań przekazuje niezwłocznie, nie później jednak niż w ciągu 15 dni od powzięcia informacji o ich wystąpieniu;
3) przekazuje do Eudravigilance Veterinary zgłoszenia pochodzące z terytorium Rzeczypospolitej Polskiej, przy czym zgłoszenia ciężkich niepożądanych działań przekazuje niezwłocznie, nie później niż w ciągu 15 dni od powzięcia informacji o ich wystąpieniu;
4) zbiera i przeprowadza ocenę raportów okresowych;
5) prowadzi bazę danych obejmującą zgłoszenia pochodzące z terytorium Rzeczypospolitej Polskiej;
6) gromadzi dane o wielkości sprzedaży produktów leczniczych weterynaryjnych na terytorium Rzeczypospolitej Polskiej pochodzące z raportów okresowych.
1) zbieranie i zestawianie informacji o wszystkich przypadkach działań niepożądanych produktów leczniczych weterynaryjnych, ze szczególnym uwzględnieniem informacji dotyczących działań, o których mowa w § 2 pkt 1;
2) ocenę bezpieczeństwa stosowania produktu leczniczego weterynaryjnego po otrzymaniu pozwolenia, w tym ocenę działania niepożądanego produktu leczniczego weterynaryjnego, ocenę niezgodnego z przeznaczeniem stosowania produktu leczniczego weterynaryjnego, ocenę doniesień wskazujących, że wyznaczony okres karencji jest niewłaściwy, ocenę toksycznego wpływu na środowisko oraz przedstawianie opracowań dotyczących bezpieczeństwa stosowania produktów leczniczych weterynaryjnych;
3) udzielenie szybkiej i pełnej odpowiedzi na każde wezwanie Prezesa Urzędu o przekazanie dodatkowych informacji niezbędnych do oceny stosunku korzyści do ryzyka oraz zagrożeń związanych ze stosowaniem produktu leczniczego weterynaryjnego;
4) niezwłoczne zgłaszanie informacji odnoszącej się do oceny stosunku korzyści do ryzyka stosowania produktu leczniczego weterynaryjnego do Prezesa Urzędu;
5) niezwłoczne informowanie Prezesa Urzędu o konieczności podjęcia natychmiastowych działań mających na celu ochronę zdrowia publicznego lub ochronę zdrowia zwierząt.
1) pochodzące ze zgłoszenia lub
2) pochodzące z literatury fachowej, lub
3) dotyczące działania niepożądanego produktu leczniczego weterynaryjnego uzyskane w wyniku prowadzenia badań dotyczących bezpieczeństwa.
1) imię i nazwisko osoby przekazującej raport w imieniu podmiotu odpowiedzialnego, wraz z jej służbowym: adresem, numerem telefonu i faksu, jeżeli dotyczy;
2) numer zgłoszenia nadany przez podmiot odpowiedzialny, jeżeli dotyczy;
3) datę otrzymania zgłoszenia przez podmiot odpowiedzialny lub osobę działającą w imieniu tego podmiotu;
4) wskazanie źródła danych, o których mowa w § 4;
5) kod umożliwiający identyfikację osoby zgłaszającej w celu uniknięcia powielenia zgłoszenia, informacje o zawodzie, jeżeli zgłoszenie obserwowanego działania niepożądanego wynika z wykonywanej pracy;
6) wskazanie kraju, w którym wystąpiło działanie niepożądane produktu leczniczego weterynaryjnego;
7) wskazanie kraju, w którym produkt leczniczy weterynaryjny został nabyty;
8) liczbę zwierząt poddanych leczeniu produktem leczniczym weterynaryjnym;
9) opis zwierząt, u których zaobserwowano objawy działania niepożądanego produktu leczniczego weterynaryjnego, z podaniem:
a) gatunku,
b) rasy,
c) płci,
d) wieku,
e) masy ciała;
10) nazwę produktu leczniczego weterynaryjnego oraz jego nazwę powszechnie stosowaną;
11) numer pozwolenia na dopuszczenie do obrotu produktu leczniczego weterynaryjnego;
12) kod międzynarodowej klasyfikacji anatomiczno-terapeutyczno-chemicznej produktów leczniczych weterynaryjnych (ATCvet);
13) postać farmaceutyczną produktu leczniczego weterynaryjnego;
14) numer serii produktu leczniczego weterynaryjnego;
15) termin ważności produktu leczniczego weterynaryjnego, jeżeli dotyczy;
16) warunki przechowywania produktu leczniczego weterynaryjnego, jeżeli dotyczy;
17) wskazanie, czy produkt zastosował lekarz weterynarii, właściciel, opiekun zwierzęcia, czy też osoba trzecia;
18) powód podania produktu leczniczego weterynaryjnego, łącznie z diagnozą;
19) zastosowaną dawkę i częstość podawania produktu leczniczego weterynaryjnego, jeżeli dotyczy;
20) drogę podania produktu leczniczego weterynaryjnego;
21) datę rozpoczęcia leczenia;
22) datę zakończenia leczenia albo długość leczenia;
23) czas pomiędzy podaniem produktu leczniczego weterynaryjnego a wystąpieniem działania niepożądanego produktu leczniczego weterynaryjnego;
24) działania podjęte po zaobserwowaniu objawów, takie jak w szczególności zmniejszenie dawki, zaprzestanie podawania;
25) informacje o wcześniej obserwowanych działaniach niepożądanych produktu leczniczego weterynaryjnego, łącznie z datą wcześniejszego leczenia, opisem obserwowanego działania niepożądanego i zakończeniem działania niepożądanego, jeżeli dotyczy;
26) informacje na temat produktu leczniczego podawanego jednocześnie z produktem, o którym mowa w pkt 10, jeżeli dotyczy, w tym:
a) nazwę tego produktu oraz jego nazwę powszechnie stosowaną,
b) numer pozwolenia na dopuszczenie do obrotu,
c) kod międzynarodowej klasyfikacji anatomiczno-terapeutyczno-chemicznej produktów leczniczych (ATC) albo produktów leczniczych weterynaryjnych (ATCvet),
d) postać farmaceutyczną,
e) numer serii produktu leczniczego,
f) termin ważności, jeżeli dotyczy,
g) warunki przechowywania, jeżeli dotyczy,
h) wskazanie, czy produkt leczniczy zastosował lekarz weterynarii, właściciel, opiekun zwierzęcia, czy też osoba trzecia,
i) drogę podania,
j) datę rozpoczęcia leczenia,
k) datę zakończenia leczenia albo długość leczenia,
l) inne informacje na temat produktu leczniczego podawanego jednocześnie;
27) informacje ma temat zaobserwowanego działania niepożądanego produktu leczniczego weterynaryjnego zawierające:
a) opis zaobserwowanego działania niepożądanego produktu leczniczego weterynaryjnego, łącznie z lokalizacją, stopniem ciężkości i objawami klinicznymi,
b) datę wystąpienia objawów,
c) datę ustąpienia objawów albo czas trwania działania niepożądanego,
d) opis leczenia albo działań podjętych po zaobserwowaniu działania niepożądanego produktu leczniczego weterynaryjnego,
e) liczbę zwierząt wykazujących objawy działania niepożądanego produktu leczniczego weterynaryjnego,
f) liczbę zwierząt padłych,
g) działania podjęte po zaobserwowaniu objawów, takie jak w szczególności zmniejszenie dawki, zaprzestanie podawania,
h) liczbę zwierząt, które przeżyły, ale nie powróciły do pełni zdrowia, jeżeli dane te są dostępne,
i) liczbę zwierząt, które przeżyły i powróciły do pełni zdrowia, jeżeli dane te są dostępne,
j) inne informacje, które mogą być pomocne w ocenie działania niepożądanego produktu leczniczego weterynaryjnego, w szczególności dotyczące podatności zwierzęcia na alergie, zmiany nawyków żywieniowych, poziomu produkcji,
k) w przypadku zwierząt padłych – związek śmierci z obserwowanym działaniem niepożądanym produktu leczniczego weterynaryjnego, z przytoczeniem wyników badań przeprowadzonych po śmierci oraz wyników badań laboratoryjnych, jeżeli takie zostały wykonane;
28) ocenę przyczynowo-skutkową obserwowanego działania niepożądanego produktu leczniczego weterynaryjnego.
2. Raport dotyczący obserwowanego u człowieka działania niepożądanego produktu leczniczego weterynaryjnego zawiera:
1) imię i nazwisko osoby przekazującej raport w imieniu podmiotu odpowiedzialnego, wraz z jej służbowym: adresem, numerem telefonu i faksu, jeżeli dotyczy;
2) numer zgłoszenia nadany przez podmiot odpowiedzialny, jeżeli dotyczy;
3) datę otrzymania zgłoszenia przez podmiot odpowiedzialny lub osobę działającą w imieniu tego podmiotu;
4) wskazanie źródła danych, o których mowa w § 4;
5) wskazanie kraju, w którym wystąpiło działanie niepożądane produktu leczniczego weterynaryjnego;
6) wskazanie kraju, w którym produkt leczniczy weterynaryjny został nabyty;
7) dane, o których mowa w art. 24 ust. 12 pkt 1 ustawy, albo kod umożliwiający identyfikację osoby zgłaszającej w celu uniknięcia powielenia zgłoszenia, informacje o zawodzie, jeżeli ma to związek z ekspozycją na produkt leczniczy weterynaryjny;
8) datę zastosowania produktu leczniczego weterynaryjnego albo datę ekspozycji na ten produkt;
9) datę wystąpienia działania niepożądanego produktu leczniczego weterynaryjnego;
10) dane produktu leczniczego weterynaryjnego: nazwę, numer pozwolenia, nazwę substancji czynnej, kod międzynarodowej klasyfikacji anatomiczno-terapeutyczno-chemicznej produktów leczniczych weterynaryjnych (ATCvet);
11) okoliczności ekspozycji, takie jak w szczególności inhalacja, wstrzyknięcie, kontakt ze skórą, typ ekspozycji oraz czas jej trwania;
12) opis zaobserwowanego działania niepożądanego produktu leczniczego weterynaryjnego, z podaniem objawów;
13) informacje o skutkach wystąpienia działania niepożądanego produktu leczniczego weterynaryjnego, takich jak w szczególności całkowite wyleczenie, konieczność dalszego leczenia;
14) nazwę (firmę), adres, numer telefonu podmiotu leczniczego udzielającego pomocy;
15) komentarz podmiotu odpowiedzialnego na temat obserwowanego działania niepożądanego;
16) informacje o leczonym zwierzęciu, takie jak droga i miejsce podania, liczbę i gatunek leczonych zwierząt i datę leczenia;
17) w przypadku gdy osoba zgłaszająca jest inna niż ta, u której wystąpiło działanie niepożądane, wskazanie, czy jest ona lekarzem weterynarii, właścicielem, opiekunem zwierzęcia, czy też osobą trzecią oraz dane tej osoby, o których mowa w art. 36e ust. 1 pkt 2–4 ustawy.
3. Dodatkowe informacje, które podmiot sporządzający raport uzyskał po jego przekazaniu, przesyła się w postaci raportu uzupełniającego.
1) nie zgadza się z oceną przyczynowo-skutkową pomiędzy zastosowaniem produktu leczniczego weterynaryjnego a jego działaniem niepożądanym, dokonaną przez osobę zgłaszającą to działanie;
2) osoba zgłaszająca działanie niepożądane produktu leczniczego weterynaryjnego nie podała własnej oceny przyczynowo-skutkowej pomiędzy zastosowaniem produktu leczniczego weterynaryjnego a wystąpieniem obserwowanego działania niepożądanego na podanie produktu leczniczego weterynaryjnego.
2. W celu ułatwienia identyfikacji zgłoszenia podmiot odpowiedzialny przekazuje całość posiadanych informacji, jeżeli jest to możliwe łącznie z numerem zgłoszenia w bazie, o której mowa w § 2 pkt 5.
2. W przypadku gdy zgłoszenie otrzymane od właściwego organu państwa członkowskiego Unii Europejskiej lub państwa członkowskiego Europejskiego Porozumienia o Wolnym Handlu (EFTA) – strony umowy o Europejskim Obszarze Gospodarczym mogłoby prowadzić do zmiany oceny stosunku korzyści do ryzyka dla danego produktu leczniczego weterynaryjnego, informację taką podmiot odpowiedzialny przekazuje niezwłocznie do Prezesa Urzędu.
1) dane dotyczące podmiotu odpowiedzialnego i produktu leczniczego weterynaryjnego, w tym:
a) nazwę (firmę) podmiotu odpowiedzialnego,
b) nazwę produktu leczniczego weterynaryjnego,
c) numer pozwolenia na dopuszczenie do obrotu,
d) europejską datę referencyjną (EBD) albo datę rozpoczęcia cyklu składania raportów okresowych,
e) informację o okresie objętym raportem okresowym,
f) datę pierwszego wprowadzenia produktu leczniczego weterynaryjnego do obrotu,
g) informację, którym w kolejności jest składany raport okresowy;
2) podsumowanie informacji o podjętych przez właściwy organ państwa członkowskiego Unii Europejskiej lub państwa członkowskiego Europejskiego Porozumienia o Wolnym Handlu (EFTA) – strony umowy o Europejskim Obszarze Gospodarczym lub podmiot odpowiedzialny działaniach związanych z bezpieczeństwem produktu leczniczego weterynaryjnego;
3) Charakterystykę Produktu Leczniczego Weterynaryjnego;
4) informacje na temat ekspozycji na produkt leczniczy weterynaryjny;
5) informacje na temat częstości występowania działań niepożądanych produktu leczniczego weterynaryjnego;
6) analizę danych pochodzących ze zgłoszeń otrzymanych w okresie objętym raportem;
7) przegląd i analizę informacji na temat bezpieczeństwa produktu leczniczego weterynaryjnego pochodzących z innych źródeł niż zgłoszenia;
8) inne informacje dotyczące bezpieczeństwa;
9) całościową ocenę danych dotyczących bezpieczeństwa produktu leczniczego weterynaryjnego;
10) informacje dotyczące bezpieczeństwa produktu leczniczego weterynaryjnego otrzymane po zakończeniu zbierania danych do przedstawianego raportu okresowego;
11) tabelaryczne zestawienie danych na temat zgłoszeń dotyczących działań niepożądanych produktu leczniczego weterynaryjnego obserwowanych u zwierząt, obejmujące następujące dane:
a) nadany przez podmiot odpowiedzialny numer zgłoszenia, zawierający kod państwa, w którym obserwowano działanie niepożądane,
b) numer zgłoszenia nadany przez właściwy organ państwa członkowskiego Unii Europejskiej lub państwa członkowskiego Europejskiego Porozumienia o Wolnym Handlu (EFTA) – strony umowy o Europejskim Obszarze Gospodarczym, jeżeli dotyczy,
c) datę leczenia lub szczepienia,
d) informacje, czy produkt był stosowany zgodnie z Charakterystyką Produktu Leczniczego Weterynaryjnego,
e) datę wystąpienia działania niepożądanego produktu leczniczego weterynaryjnego,
f) liczbę, gatunek i wiek leczonych zwierząt,
g) liczbę zwierząt (przybliżoną), u których obserwowano działanie niepożądane, jeżeli dotyczy,
h) liczbę zwierząt padłych,
i) inne produkty podawane jednocześnie, włącznie z premiksami (nazwa i substancja czynna),
j) obserwowane objawy działania niepożądanego lub rozpoznanie włącznie z czasem ich trwania,
k) terminologię VEDDRA opisującą obserwowane objawy działania niepożądanego lub rozpoznanie,
l) krótki komentarz podmiotu odpowiedzialnego,
m) ocenę przyczynowo-skutkową;
12) tabelaryczne zestawienie danych na temat zgłoszeń dotyczących działań niepożądanych produktu leczniczego weterynaryjnego obserwowanych u człowieka, obejmujące następujące dane:
a) nadany przez podmiot odpowiedzialny numer zgłoszenia zawierający kod państwa, w którym obserwowano działanie niepożądane,
b) numer zgłoszenia nadany przez właściwy organ państwa członkowskiego Unii Europejskiej lub państwa członkowskiego Europejskiego Porozumienia o Wolnym Handlu (EFTA) – strony umowy o Europejskim Obszarze Gospodarczym, jeżeli dotyczy,
c) datę ekspozycji na produkt leczniczy weterynaryjny,
d) datę wystąpienia objawów,
e) dane, o których mowa w art. 24 ust. 12 pkt 1 ustawy, albo kod umożliwiający identyfikację osoby zgłaszającej w celu uniknięcia powielenia zgłoszenia, informacje o zawodzie, jeżeli ma to związek z ekspozycją na produkt leczniczy weterynaryjny,
f) zawód osoby, u której obserwowano objawy działania niepożądanego, jeżeli ma to związek z ekspozycją na produkt leczniczy weterynaryjny,
g) rodzaj ekspozycji,
h) obserwowane objawy działania niepożądanego,
i) informacje na temat ustąpienia objawów działania niepożądanego,
j) krótki komentarz podmiotu odpowiedzialnego.
2. Informacje, o których mowa w ust. 1 pkt 11 i 12, sporządza się na formularzach opracowanych przez podmiot odpowiedzialny, jeżeli zawierają one dane, o których mowa w ust. 1 pkt 11 i 12, zgodnie z wiążącymi wytycznymi, o których mowa w art. 77 dyrektywy 2001/82/WE Parlamentu Europejskiego i Rady z dnia 6 listopada 2001 r. w sprawie wspólnotowego kodeksu odnoszącego się do weterynaryjnych produktów leczniczych.
2. Informacje o bezpieczeństwie produktów leczniczych weterynaryjnych złożonych, które zawierają substancje czynne będące przedmiotem odrębnych raportów okresowych, podmiot odpowiedzialny może przedstawić w osobnym raporcie okresowym lub włączyć jako odrębne opracowanie do raportu okresowego dla produktu leczniczego weterynaryjnego zawierającego jedną z substancji czynnych.
Minister Zdrowia: B.A. Arłukowicz
|
|
1) Minister Zdrowia kieruje działem administracji rządowej - zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 18 listopada 2011 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 248, poz. 1495 i Nr 284, poz. 1672).
2) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2008 r. Nr 227, poz. 1505 i Nr 234, poz. 1570, z 2009 r. Nr 18, poz. 97, Nr 31, poz. 206, Nr 92, poz. 753, Nr 95, poz. 788 i Nr 98, poz. 817, z 2010 r. Nr 78, poz. 513 i Nr 107, poz. 679, z 2011 r. Nr 63, poz. 322, Nr 82, poz. 451, Nr 106, poz. 622, Nr 112, poz. 654, Nr 113, poz. 657 i Nr 122, poz. 696, z 2012 r. poz. 1342 i 1544 oraz z 2013 r. poz. 1245.
3) Niniejsze rozporządzenie było poprzedzone rozporządzeniem Ministra Zdrowia z dnia 17 lutego 2003 r. w sprawie monitorowania bezpieczeństwa produktów leczniczych (Dz. U. Nr 47, poz. 405), które utraciło moc z dniem wejścia w życie ustawy z dnia 27 września 2013 r. o zmianie ustawy - Prawo farmaceutyczne oraz niektórych innych ustaw (Dz. U. poz. 1245).
Załącznik do rozporządzenia Ministra Zdrowia
z dnia 3 czerwca 2014 r. (poz. 806)
WZÓR – FORMULARZ ZGŁOSZENIA PODEJRZENIA POJEDYNCZEGO PRZYPADKU DZIAŁANIA NIEPOŻĄDANEGO PRODUKTU LECZNICZEGO WETERYNARYJNEGO


- Data ogłoszenia: 2014-06-18
- Data wejścia w życie: 2014-07-03
- Data obowiązywania: 2014-07-03

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA