REKLAMA
Dziennik Ustaw - rok 2010 nr 202 poz. 1341
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 18 października 2010 r.
w sprawie sposobu dokonywania zgłoszeń i powiadomień dotyczących wyrobów
Na podstawie art. 65 ust. 2 ustawy z dnia 20 maja 2010 r. o wyrobach medycznych (Dz. U. Nr 107, poz. 679) zarządza się, co następuje:
1) wzory formularzy zgłoszeń i powiadomień, o których mowa w art. 58 i art. 61 ustawy z dnia 20 maja 2010 r. o wyrobach medycznych, zwanej dalej „ustawą”;
2) sposób zgłaszania zmiany danych objętych zgłoszeniem i powiadomieniem;
3) sposób zgłaszania zaprzestania wprowadzania wyrobu do obrotu oraz zaprzestania pełnienia funkcji autoryzowanego przedstawiciela;
4) sposób przekazywania Prezesowi Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych formularzy oraz dokumentów dołączanych do zgłoszenia lub powiadomienia.
2. Wzór formularza zgłoszenia danych dotyczących wyrobów medycznych do diagnostyki in vitro, w tym wyrobów do oceny działania, lub powiadomienia o rozpoczęciu na terytorium Rzeczypospolitej Polskiej oceny działania wyrobów do oceny działania jest określony w załączniku nr 3 do rozporządzenia.
3. Wzór formularza powiadomienia przez dystrybutora albo importera o wprowadzeniu na terytorium Rzeczypospolitej Polskiej aktywnych wyrobów medycznych do implantacji, wyrobów medycznych, systemów lub zestawów zabiegowych lub wyrobów medycznych do diagnostyki in vitro jest określony w załączniku nr 4 do rozporządzenia.
1) formularz, o którym mowa w § 2;
2) odpowiednią liczbę formularzy, o których mowa w § 3 ust. 1; dla każdego wyrobu danego wytwórcy wypełnia się jeden formularz.
2. W celu dokonania zgłoszenia wyrobu medycznego do diagnostyki in vitro, w tym wyrobu do oceny działania, podmiot dokonujący zgłoszenia wypełnia:
1) formularz, o którym mowa w § 2;
2) odpowiednią liczbę formularzy, o których mowa w § 3 ust. 2; dla każdego wyrobu danego wytwórcy wypełnia się jeden formularz.
3. W celu dokonania zgłoszenia systemu lub zestawu zabiegowego albo działalności polegającej na sterylizacji systemów, zestawów zabiegowych lub wyrobów medycznych oznakowanych znakiem CE podmiot dokonujący zgłoszenia wypełnia:
1) formularz, o którym mowa w § 2;
2) odpowiednią liczbę formularzy, o których mowa w § 3 ust. 1; dla każdego systemu lub zestawu zabiegowego – zestawianego lub sterylizowanego, i dla sterylizowanego wyrobu medycznego wypełnia się jeden formularz.
4. W celu dokonania powiadomienia o wprowadzeniu na terytorium Rzeczypospolitej Polskiej aktywnego wyrobu medycznego do implantacji, wyrobu medycznego, systemu lub zestawu zabiegowego lub wyrobu medycznego do diagnostyki in vitro podmiot dokonujący powiadomienia wypełnia:
1) formularz, o którym mowa w § 2; dla każdego wytwórcy, o którego wyrobach podmiot będzie powiadamiał, wypełnia się jeden formularz;
2) odpowiednią liczbę formularzy, o których mowa w § 3 ust. 3; dla każdego kolejnego wytwórcy wypełnia się osobne formularze zawierające informacje o maksymalnie 20 wyrobach.
5. W celu dokonania powiadomienia o rozpoczęciu wykonywania na terytorium Rzeczypospolitej Polskiej oceny działania wyrobu do oceny działania świadczeniodawca dokonujący powiadomienia wypełnia:
1) formularz, o którym mowa w § 2;
2) odpowiednią liczbę formularzy, o których mowa w § 3 ust. 2; dla każdego wyrobu do oceny działania wypełnia się jeden formularz.
1) adresu podmiotu (ulica, numer domu lub lokalu, miejscowość i kod pocztowy).
2) nazwy podmiotu,
3) osoby do kontaktu
– podmiot wypełnia formularz, o którym mowa w § 2.
2. W celu zmiany danych objętych zgłoszeniem związanych z wyrobem, dotyczących:
1) numeru jednostki notyfikowanej,
2) nazwy handlowej lub nazwy rodzajowej wyrobu,
3) klasy wyrobu medycznego albo kwalifikacji wyrobu medycznego do diagnostyki in vitro,
4) kodu według Globalnej Nomenklatury Wyrobów Medycznych (GMDN) albo innej uznanej nomenklatury wyrobów medycznych
– podmiot wypełnia formularz, o którym mowa w § 2, oraz odpowiednią liczbę formularzy, o których mowa w § 3 ust. 1 lub 2; dla każdego wyrobu danego wytwórcy wypełnia się jeden formularz.
3. W celu zmiany danych objętych powiadomieniem, dotyczących nazwy lub adresu dystrybutora, importera lub świadczeniodawcy wykonującego ocenę działania wyrobu do oceny działania, podmiot wypełnia formularz, o którym mowa w § 2.
4. W celu zmiany danych objętych powiadomieniem, dotyczących nazwy lub adresu wytwórcy lub autoryzowanego przedstawiciela, lub nazwy handlowej wyrobu, podmiot wypełnia:
1) formularz, o którym mowa w § 2; dla każdego wytwórcy lub autoryzowanego przedstawiciela, którego dotyczą zgłaszane zmiany, wypełnia się jeden formularz;
2) odpowiednią liczbę formularzy, o których mowa w § 3 ust. 3; dla każdego kolejnego wytwórcy lub autoryzowanego przedstawiciela wypełnia się osobne formularze zawierające informacje o maksymalnie 20 wyrobach.
5. Do wniosku o zmianę danych podmiot dołącza dokumenty dotyczące wyrobu, o których mowa w art. 59 ust. 2 i art. 60 ust. 2 ustawy, których treść uległa zmianie.
6. W celu dokonania zmiany danych osoby do kontaktu lub osoby do kontaktu w sprawach incydentów medycznych podmiot wypełnia:
1) formularz, o którym mowa w § 2 – w przypadku zmiany danych osoby do kontaktu, która jest jednocześnie osobą do kontaktu w sprawach incydentów medycznych dla wszystkich wyrobów danego wytwórcy;
2) formularz, o którym mowa w § 2, oraz odpowiednio formularz, o którym mowa w § 3 ust. 1 lub 2 – w pozostałych przypadkach.
1) datę zaprzestania;
2) nazwę i adres wytwórcy wyrobu;
3) nazwę handlową wyrobu;
4) datę sporządzenia dokumentu i podpis osoby odpowiedzialnej.
2. Podmiot zgłasza w formie pisemnej fakt zaprzestania pełnienia funkcji autoryzowanego przedstawiciela, podając:
1) datę zaprzestania;
2) nazwę i adres autoryzowanego przedstawiciela;
3) nazwę i adres wytwórcy wyrobu, którego reprezentował jako autoryzowany przedstawiciel;
4) nazwę handlową wyrobu;
5) datę sporządzenia dokumentu i podpis osoby odpowiedzialnej.
2. Papierową wersję zgłoszenia, powiadomienia lub zgłoszenia zmiany danych sporządza się, wypełniając interaktywne formularze pobrane ze strony internetowej Urzędu Rejestracji i dokonując ich wydruku. Dokument elektroniczny sporządza się, zapisując dane z wypełnionych formularzy bezpośrednio po ich wydrukowaniu.
3. Dokumentację towarzyszącą zgłoszeniu lub powiadomieniu składa się w wersji papierowej lub w postaci dokumentu elektronicznego, która może obejmować wzory:
1) oznakowania wyrobu;
2) instrukcji używania;
3) materiałów promocyjnych.
4. Dokument elektroniczny zgłoszenia, powiadomienia lub zgłoszenia zmiany danych oraz dokumentacji towarzyszącej przekazuje się w postaci plików zapisanych na nośniku optycznym (CD lub DVD) do jednorazowego zapisu.
Minister Zdrowia: E. Kopacz
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 16 listopada 2007 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 216, poz. 1607).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 18 października 2010 r. (poz. 1341)
Załącznik nr 1
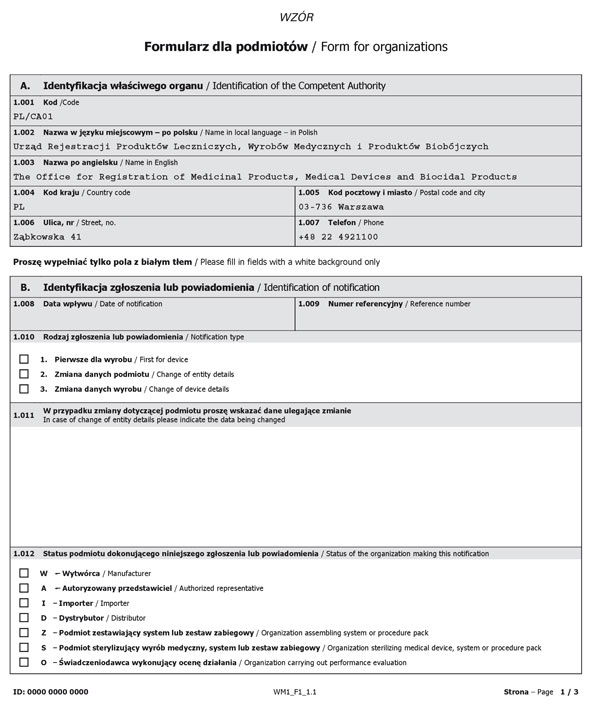
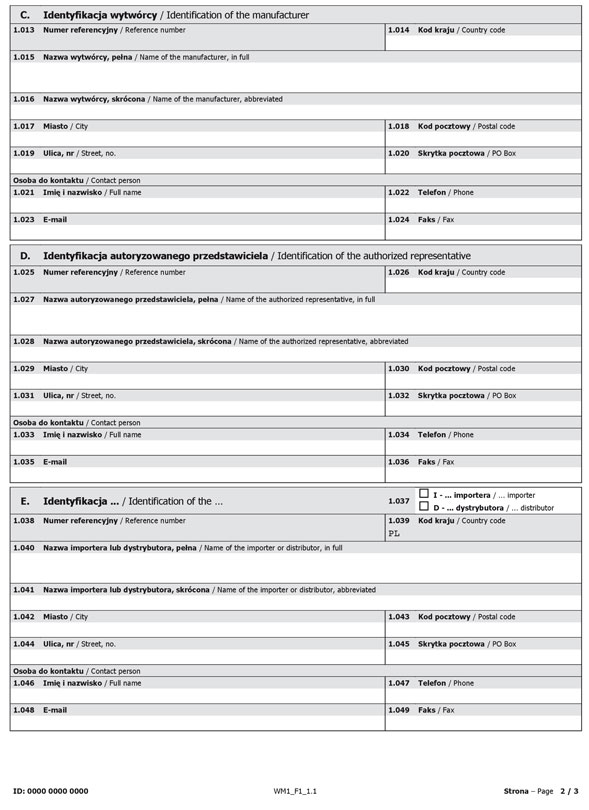
Formularz dla podmiotów

Załącznik nr 2
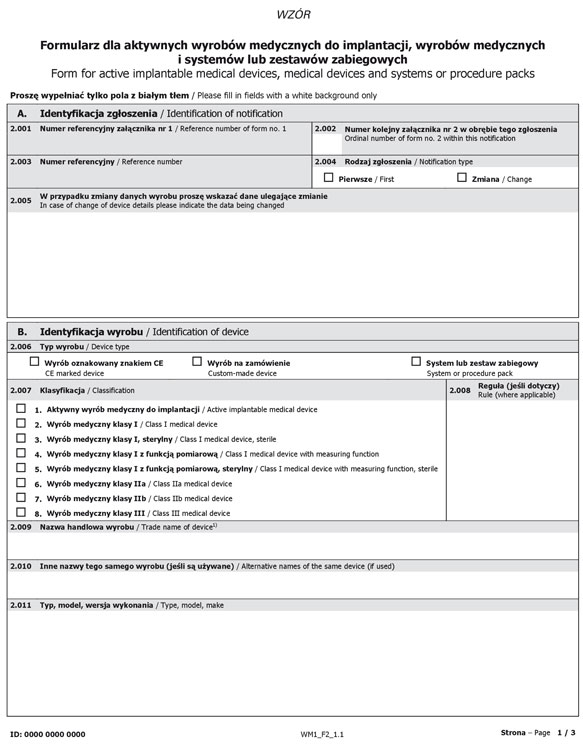
Formularz dla aktywnych wyrobów medycznych do implantacji, wyrobów medycznych i systemów lub zestawów zabiegowych


Załącznik nr 3
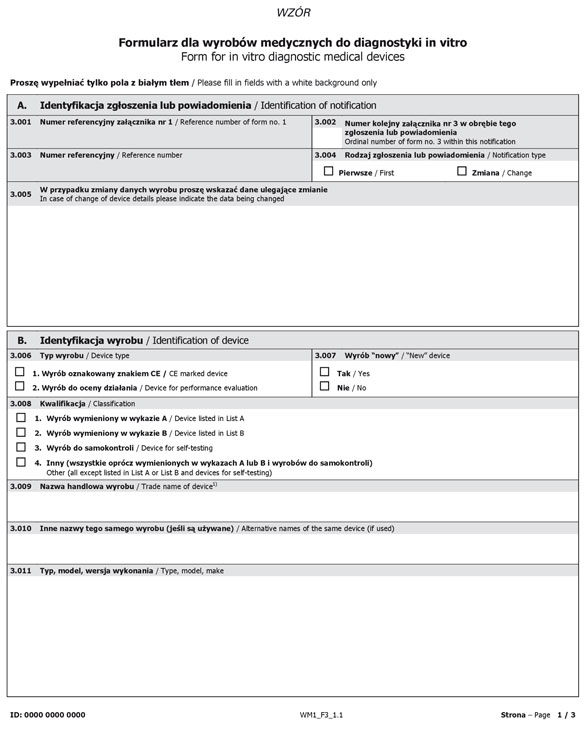
Formularz dla wyrobów medycznych do diagnostyki in vitro


Załącznik nr 4
Wykaz wyrobów objętych powiadomieniem

- Data ogłoszenia: 2010-10-29
- Data wejścia w życie: 2010-10-29
- Data obowiązywania: 2014-10-23
- Dokument traci ważność: 2016-01-12

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA