REKLAMA
Dziennik Ustaw - rok 2003 nr 9 poz. 107
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 23 grudnia 2002 r.
w sprawie określenia procedur pobierania próbek kosmetyków oraz procedur przeprowadzania badań laboratoryjnych
Na podstawie art. 13 ust. 3 ustawy z dnia 30 marca 2001 r. o kosmetykach (Dz. U. Nr 42, poz. 473) zarządza się, co następuje:
1) procedury pobierania próbek kosmetyków;
2) procedury przeprowadzania badań laboratoryjnych.
1) w celu dokonania rutynowej kontroli lub
2) w związku z uzasadnionym podejrzeniem, że kosmetyk nie spełnia wymagań jakościowych deklarowanych przez producenta, lub
3) w związku z uzasadnionym podejrzeniem, że kosmetyk może szkodzić zdrowiu ludzi lub środowisku.
2. Próbki pobiera się w ilościach niezbędnych do przeprowadzenia badań laboratoryjnych.
1) pieczęć organu Państwowej Inspekcji Sanitarnej;
2) numer protokołu;
3) imię, nazwisko i stanowisko służbowe pobierającego próbkę oraz numer upoważnienia do przeprowadzenia kontroli sanitarnej;
4) datę i miejsce pobrania próbki;
5) warunki, w jakich kosmetyk był przechowywany;
6) opis sposobu pobrania i zabezpieczenia próbki;
7) imię i nazwisko lub nazwę (firmę) oraz adres lub siedzibę producenta;
8) nazwę, termin trwałości i numer serii kosmetyku, którego próbkę pobrano, jeżeli numer taki znajduje się na oznakowaniu opakowania jednostkowego;
9) cenę kosmetyku, którego próbkę pobrano;
10) określenie proponowanego zakresu badań laboratoryjnych;
11) własnoręczne podpisy pobierającego próbkę oraz producenta lub osoby przez niego upoważnionej.
2. Protokół sporządza się w dwóch egzemplarzach, z których jeden pozostawia się – za pokwitowaniem – kontrolowanemu producentowi, drugi pozostawia się w aktach z przeprowadzonej kontroli sanitarnej.
2. Dodatkowa próbka kosmetyku powinna być przechowywana przez kontrolowanego, z zachowaniem warunków uniemożliwiających zmianę jakości lub cech charakterystycznych kosmetyku, i do czasu ustalenia wyników badań laboratoryjnych nie może być wprowadzona do obrotu.
3. Organ Państwowej Inspekcji Sanitarnej powiadamia producenta o wynikach badań laboratoryjnych.
2. Dokumentację dotyczącą badań, o których mowa w ust. 1, prowadzi podmiot przeprowadzający badania w sposób określony w załączniku nr 1 do rozporządzenia.
3. Organ Państwowej Inspekcji Sanitarnej sporządza protokół przeprowadzonych badań, zawierający dane określone w § 3 ust. 1 pkt 1, 2, 5, 7 i 8, a ponadto:
1) datę i miejsce wykonania badania;
2) imię, nazwisko i stanowisko służbowe przeprowadzającego badania;
3) opis sposobu pobrania próbki laboratoryjnej i stosowanych metod badania;
4) ocenę prawidłowości oznakowania kosmetyku;
5) wyniki badania;
6) własnoręczny podpis przeprowadzającego badanie.
4. Do protokołu, o którym mowa w ust. 3, stosuje się odpowiednio § 3 ust. 2.
Minister Zdrowia: M. Łapiński
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 28 czerwca 2002 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 93, poz. 833).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 23 grudnia 2002 r. (poz. 107)
Załącznik nr 1
KRYTERIA CZYSTOŚCI CHEMICZNEJ MIKROBIOLOGICZNEJ KOSMETYKÓW ORAZ METODY KONTROLI ZGODNOŚCI Z TYMI KRYTERIAMI
1. Opracowywanie nowego kosmetyku
1. 1. Konserwowanie wyrobu
Zadaniem środków konserwujących (ŚK) jest ochrona konsumenta i zapobieganie zanieczyszczeniom mikrobiologicznym w czasie normalnego i możliwego do przewidzenia stosowania kosmetyku. ŚK nie powinny być stosowane zamiast utrzymania na właściwym poziomie higieny produkcji. Kontrola czystości mikrobiologicznej w czasie produkcji jest osiągana poprzez zrozumienie przyczyn występowania zanieczyszczeń i ich eliminowanie.
Należy podkreślić, że ŚK nie mogą być dobierane wyłącznie o dane teoretyczne, ale konieczne jest określenie in situ ich efektywności poprzez stosowanie testów kontrolowanego zanieczyszczenia mikrobiologicznego wyrobów w trakcie ich opracowywania.
1. 2. Opracowywanie prawidłowych receptur
W miarę możliwości powinno się tworzyć receptury, w których zastosowane surowce nie wspomagają wzrostu drobnoustrojów, dzięki czemu można ograniczyć potrzebę dodawania ŚK. Jeśli jednak użycie ŚK jest konieczne, powinien on być wybrany na początku opracowywania wyrobu i traktowany jako integralna część receptury.
Woda jest podstawą wzrostu mikroorganizmów. Układ konserwujący powinien być tak dobrany, aby w fazie wodnej systemu wielofazowego uzyskać stężenie efektywne.
Układ konserwujący powinien być skuteczny dla szerokiego zakresu mikroorganizmów i bezpieczny w użytym stężeniu. Mieszaniny ŚK mogą być czasem bardziej efektywne niż pojedyncze składniki. Bardzo ważne są temperatura, światło i stabilność podczas przedłużonego okresu przechowywania. Układ konserwujący powinien być skuteczny przy niskim stężeniu i pH wyrobu oraz wykazywać zgodność z innymi składnikami receptury.
Zaprojektowane opakowanie powinno uniemożliwiać dostęp drobnoustrojów i wydzielania wody z produktu, co może ułatwić wzrost drobnoustrojów. Należy wziąć pod uwagę możliwość inaktywa-cji układu konserwującego przez składniki opakowania i dyfuzję przez opakowanie.
Użycie konserwantów jest szczególnie ważne przy zastosowaniu łatwo biodegradowalnych surowców w recepturach, ponieważ surowce takie są składnikami odżywczymi dla drobnoustrojów.
Każda zmiana składu chemicznego może wpłynąć na skuteczność ŚK. W takich przypadkach należy rozważyć przeprowadzenie dodatkowych testów.
Podczas produkcji niektórych kosmetyków może być konieczne przygotowywanie mieszanin wstępnych. Wstępne wodne mieszaniny powinny zawierać konserwanty i powinny być przechowywane przez określony czas i regularnie kontrolowane. Aby określić ich podatność na zanieczyszczenia mikrobiologiczne, powinny być poddane testom kontrolowanego zanieczyszczenia mikrobiologicznego.
1. 3 Testy kontrolowanego zanieczyszczenia mikrobiologicznego
Jeżeli jest konieczne przeprowadzenie testu kontrolowanego zanieczyszczenia mikrobiologicznego, powinien być wykonany przez wykwalifikowaną osobę z użyciem kompletnej receptury wyrobu w opakowaniu handlowym, nawet jeśli prowadzone były wstępne badania tej receptury podczas opracowywania kosmetyku.
Zasadą jest wprowadzanie do wyrobu różnych mikroorganizmów: bakterii, drożdży, pleśni, wśród których mogą być użyte drobnoustroje chorobotwórcze lub powodujące zepsucie kosmetyku. W tak zanieczyszczonym produkcie badana jest przeżywalność mikroorganizmów w różnych okresach – tak długo, jak uważa się to za konieczne.
2. Surowce
Surowce mogą być źródłem zanieczyszczenia mikroorganizmami wyrobu gotowego. Celem producentów surowców powinno być zatem dostarczanie ich o niskim stopniu zanieczyszczeń i wolnych od szkodliwych drobnoustrojów.
Woda jest jednym z głównych surowców kosmetycznych i w określonych warunkach może być znacznie zanieczyszczona mikrobiologicznie, co może stwarzać zagrożenie dla trwałości gotowego wyrobu. Dlatego też stanowiącą składnik receptur wodę należy regularnie kontrolować i gdy to konieczne – odpowiednio przygotować.
3. Kontrola i ocena półproduktu oraz gotowego wyrobu
3. 1. Przechowywanie partii kosmetyku przed zapakowaniem
Wyrób w „masie” jest bardziej podatny na zanieczyszczenia mikrobiologiczne niż w postaci gotowego wyrobu w opakowaniu.
3. 2. Pobieranie próbek
Z partii wyrobu powinny być pobierane próbki w momencie właściwym dla aktywności mikrobiologicznej receptury, zgodnie z ocenami dokonanymi w testach kontrolowanego zanieczyszczenia mikrobiologicznego i doświadczeniem produkcyjnym. Dla każdego nowego wyrobu należy określić częstotliwość badań w okresie początkowym, przynajmniej przez trzy miesiące, aby rozpoznać jakość mikrobiologiczną wyrobu w zwykłych warunkach produkcyjnych.
4. Dobra praktyka produkcyjna
Aby spełnić warunki Dobrej Praktyki Produkcyjnej, należy stosować Zalecenia Dobrej Praktyki w Produkcji Kosmetyków opracowanych przez stowarzyszenie COLIPA (THE EUROPEAN COSMETIC TOILETRY AND PERFUMERY ASSOCIATION).
5. Dokumentacja
Powinny być prowadzone odpowiednie zapisy odnośnie do wszystkich aspektów badań mikrobiologicznych w czasie opracowywania i produkcji każdego wyrobu oraz wszystkich procedur kontrolnych stosowanych w zakładzie produkcyjnym.
6. Zalecane wymagania czystości mikrobiologicznej i metody oceny gotowego wyrobu
6. 1. Uwagi ogólne
Celem wszystkich producentów musi być zagwarantowanie bezpieczeństwa wyrobów w zwykłych i możliwych do przewidzenia warunkach stosowania.
Zanieczyszczenia mikrobiologiczne mogą okazać się szkodliwe dla konsumenta i powodować zepsucie wyrobu, dlatego należy je ograniczać poprzez:
a) zachowanie higieny w zakładzie i procesie produkcyjnym,
b) odpowiednie zakonserwowanie wyrobu zapobiegające wzrostowi drobnoustrojów,
c) przestrzeganie granic czystości mikrobiologicznej w celu zapewnienia bezpieczeństwa wyrobu.
Podczas wykonywania pojedynczych badań producent musi pamiętać, że zanieczyszczające mikroorganizmy mogą być w fazie namnażania, statycznej lub zamierania. Użycie ŚK ma na celu niedopuszczenie do wzrostu drobnoustrojów. ŚK nie zastąpi dobra higiena w zakładzie produkcyjnym, trzeba również pamiętać, że użyte stężenie tych związków jest wypadkową między aktywnością przeciwdrobnoustrojową a bezpieczeństwem użytkownika. ŚK powinny być wybierane podczas opracowywania produktu, przy zastosowaniu testów kontrolowanego zanieczyszczenia mikrobiologicznego.
Opisana niżej metodyka powinna być traktowana jako zalecana, producenci zaś mogą stosować własne metody kontroli wewnętrznej zapewniające produkowanie wyrobów zgodnych z kryteriami zawartymi w niniejszym załączniku.
Specyfikacja mikrobiologiczna odnosi się do środków kosmetycznych w nienaruszonym, zamkniętym opakowaniu.
Wyniki badań powinny być zapisywane w raporcie, który powinien zawierać następujące informacje, dotyczące:
a) identyfikacji próbek:
– rodzaj wyrobu,
– nazwę handlową,
– nazwę producenta,
– numer partii,
– datę i miejsce pobrania próbki,
– metody identyfikacji i warunki przechowywania,
b) warunków badań:
– datę,
– metodę,
– podłoże neutralizujące,
c) wyników testów.
Raport powinien zawierać wniosek oraz dane osoby odpowiedzialnej za badania.
6. 2. Wymagania mikrobiologiczne
6. 2. 1. Wymagania ilościowe (minimalna wielkość badanej próbki – 1 g lub 1 ml)
KATEGORIA l. Kosmetyki przeznaczone dla dzieci i w okolice oczu
Ogólna liczba drobnoustrojów tlenowych mezofilnych nie może przekraczać 100 jtk/g lub ml (jtk – jednostki tworzące kolonie).
KATEGORIA II. Inne kosmetyki
Ogólna liczna drobnoustrojów tlenowych mezofilnych nie może przekraczać 1000 jtk/g lub ml.
Bardzo ważna jest interpretacja wyników. Opisane ograniczenia powinny być interpretowane następująco:
102 – maksymalny limit akceptacji to 5 x 102,
103 – maksymalny limit akceptacji to 5 x 103.
6. 2. 2. Wymagania jakościowe
W 0,1 g lub 0,1 ml próbki kosmetyku nie mogą być obecne następujące drobnoustroje:
– Pseudomonas aeruginosa,
– Staphylococcus aureus,
– Candida albicans.
6. 2. 3. Kryteria akceptacji
Akceptowana jest jakość próbek, które spełniają wymagania jakościowe i ilościowe.
6. 3. Metody badań mikrobiologicznych środków kosmetycznych
Określenie ogólnej liczby żywych drobnoustrojów tlenowych mezofilnych powinno być prowadzone według następujących zasad:
a) należy zachować ostrożność i unikać zanieczyszczenia produktu drobnoustrojami, które mogą być wykryte w próbce,
b) eliminacja każdej przeciwdrobnoustrojowej właściwości produktu w czasie analizy powinna być dokonana przez rozcieńczenie, zobojętnienie lub filtrację,
c) określenie ogólnej liczby żywych drobnoustrojów tlenowych mezofilnych prowadzi się, stosując techniki wylewania płytek, techniki posiewów powierzchniowych lub filtracji. „Wzbogacone” techniki nie są konieczne,
d) identyfikacja określonych mikroorganizmów jest prowadzona przy użyciu selektywnych podłoży hodowlanych.
6. 3. 1. Materiały i odczynniki
a) podłoża hodowlane i odczynniki,
b) przygotowanie próbki.
Ocena mikrobiologiczna powinna być przeprowadzona na próbkach wielkości minimum 1 g lub 1 ml.
Dla produktów o wadze mniejszej niż 1 g należy połączyć kilka próbek w celu otrzymania 1 g lub 1 ml.
Należy rozcieńczyć lub rozpuścić 1 ml lub 1 g wyrobu w mianowanym rozcieńczalniku neutralizującym w celu otrzymania rozcieńczenia 1:10. W przypadku słabo wilgotniejących substancji można dodać odpowiedni związek powierzchniowo czynny, np. 0,1% (m/v) polisorbatu 80.
Jeżeli konieczne jest uzyskanie kolejnych dziesiętnych rozcieńczeń, należy użyć roztworu wyjściowego, stosując ten sam rozcieńczalnik, aby osłabić aktywność przeciwdrobnoustrojową produktu lub uzyskać liczbę kolonii od 10 do 100 w celu łatwego zliczenia.
6. 3. 2. Określenie całkowitej liczby tlenowych drobnoustrojów mezofilnych
a) Filtracja przez błony
Należy stosować 2 błony filtracyjne o średnicy porów nie większej niż 0,45 µ i których efektywność w zatrzymywaniu bakterii została ustalona. Niżej opisana metoda zakłada stosowanie błon o średnicy ok. 50 mm:
– na każdą błonę przenieść ilość odpowiadającą 0,1 g badanego produktu i natychmiast przesączyć. W celu uzyskania równomiernego rozkładu drobnoustrojów na błonie, można ją przepłukać sterylnym roztworem płuczącym,
– przenieść błony na powierzchnię odpowiedniego podłoża agarowego, inkubować w 30–35°C przez 3 dni w przypadku bakterii, 20–25°C przez 5 dni w przypadku grzybów, chyba że bardziej wiarygodne zliczenia są osiągalne w krótszym czasie,
– policzyć liczbę wyhodowanych kolonii i przeliczyć liczbę mikroorganizmów na gram lub mi-lilitr badanego wyrobu.
b) Procedura liczenia płytek:
– stosując płytki Petriego o średnicy 9–10 cm, dodać do każdej płytki 1 ml próbki odpowiadającej 0,1 g wyrobu i ok. 15 ml odpowiedniego płynnego podłoża agarowego o temperaturze nie wyższej niż 45°C, wymieszać i pozostawić na chłodnej poziomej powierzchni do zastygnięcia,
– alternatywnie: rozprowadzić rozcieńczony wyrób (zwykle 0,1 ml na płytkę, co odpowiada 0,01 g wyrobu) na powierzchni stałego podłoża na płytce Petriego o średnicy j.w.,
– inkubować płytki jak w przypadku metody filtracyjnej,
– policzyć wyhodowane kolonie i przeliczyć wyniki, używając płytek z największą liczbą kolonii, lecz nie większą niż 300 kolonii dla bakterii i 100 kolonii dla grzybów.
c) Przedstawienie wyników
Należy policzyć i zapisać liczbę jednostek tworzących kolonie (jtk) dla każdej płytki. Wyliczyć całkowitą liczbę jtk/płytkę i przemnożyć wynik przez rozcieńczenie w celu uzyskania ilości jtk/g lub jtk/ml wyrobu.
6. 3. 3. Wykrywanie określonych drobnoustrojów
Specyficzne drobnoustroje:
a. Pseudomonas aeruginosa
Na podłożu z cetrimidem (pkt 7 – Roztwory i podłoża hodowlane) typowe kolonie P. aeruginosa są płaskie, przejrzyste, żółto-zielonkawe do niebieskich.
Należy przeprowadzić co najmniej następujące testy potwierdzające:
– barwienie metodą Gramma,
– test oksydazowy,
– test na ruchliwość,
– wzrost w 42°C.
P. aeruginosa jest Gram-ujemną pałeczką, ruchliwą, oksydazo-dodatnią i rośnie w temp. 42°C.
b. Staphylococcus aureus
Na podłożu Baird-Parkera (pkt 7 – Roztwory i podłoża hodowlane) typowe kolonie S. aureus są czarne, błyszczące, wypukłe, otoczone przejrzystym obszarem, który może opalizować.
Należy przeprowadzić co najmniej następujące testy potwierdzające:
– barwienie metodą Gramma,
– test katalazowy,
– test koagulazowy.
Gram-dodatnie ziarniaki, katalazo- i koagulazo-dodatnie mogą być uważane za S. aureus.
c. Candida alblcans
Na podłożu Sabouard-dekstrozowym (pkt 7 – Roztwory i podłoża hodowlane) typowe kolonie C. albicans są białe do beżowych, kremowe, wypukłe.
Należy przeprowadzić co najmniej następujące testy potwierdzające:
– badanie mikroskopowe,
– test tworzenia nibystrzępek,
– tworzenie chlamidosporów.
Drożdże tworzące nibystrzępki i dające chlamidospory mogą być uważane za C. albicans.
W każdym przypadku powinny być prowadzone inne odpowiednie testy, aby potwierdzić identyfikację.
Przedstawienie wyników:
Wyniki należy zapisać w raporcie z badań jako obecność lub nieobecność swoistych mikroorganizmów:
Pseudomonas aeruginosa, Staphylococcus aureus, Candida albicans.
7. Roztwory i podłoża hodowlane
Poniższe roztwory i podłoża hodowlane powinny być przeznaczone do stosowania zgodnie z przeznaczeniem, jednakże można stosować odpowiednie podłoża suche, pod warunkiem że mają one identyczne właściwości odżywcze i selektywne dla testowych drobnoustrojów.
7. 1. Rozcieńczalnik
Buforowany roztwór chlorku sodu z peptonem o pH 7,0.
| Dwuwodorofosforan potasu | 3,56 g |
| Wodorofosforan sodu | 7,23 g |
| Chlorek sodu | 4,30 g |
| Pepton (mięsny lub kazeinowy) | 1,0 g |
| Woda destylowana | 1000 ml |
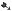
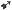
Wyjaławiać w autoklawie w 121°C przez 15 minut.
W razie konieczności można dodać odpowiednie czynniki zobojętniające, np. polisorbat 20 lub 80, lecytynę, tiosiarczan.
7. 2. Podłoże do określania liczby bakterii
Agar z hydrolizatem kazeinowo-sojowym.
| Trzustkowy hydrolizat kazeiny | 15,0 g |
| Papainowy hydrolizat ziaren soi | 5,0 g |
| Chlorek sodu | 5,0 g |
| Agar | 15,0 g |
| Woda destylowana | 1 000 ml |
Wyjaławiać 15 minut w autoklawie w temperaturze 121°C; pH po wyjałowieniu powinno wynosić 7,3±0,2.
7. 3. Podłoże do określania liczby grzybów
Pożywka Sabouard-dekstrozowa.
| Peptony (mięsny i kazeinowy) | 10,0 g |
| Dekstroza jednowodna | 40,0 g |
| Agar | 15,0 g |
| Woda destylowana | 1 000 ml |
Wyjaławiać 15 minut w autoklawie w temperaturze 121°C, pH po wyjałowieniu powinno wynosić 5,6±0,2.
7. 4. Podłoże do wykrywania Pseudomonas aeruginosa
Pożywka z cetrimidem.
| Trzustkowy hydrolizat żelatyny | 20,0 g |
| Chlorek magnezu | 1,4 g |
| Siarczan potasu | 10,0 g |
| Cetrimid | 0,3 g |
| Agar | 13,6 g |
| Glicerol | 10,0 ml |
| Woda destylowana | 1 000 ml |
Wyjaławiać 15 minut w autoklawie w temperaturze 121°C; pH po wyjałowieniu powinno wynosić 7,2±0,2.
7. 5. Podłoże do wykrywania Staphylococcus aureus
Pożywka Baird-Parkera.
| Trzustkowy hydrolizat kazeiny | 10,0 g |
| Wyciąg wołowy | 5,0 g |
| Wyciąg drożdżowy | 1,0 g |
| Chlorek litu | 5,0 g |
| Agar | 20,0 g |
| Glicyna | 12,0 g |
| Pirogronian sodu | x10,0g |
| Woda destylowana | 950 ml |
Po wyjałowieniu pH powinno wynosić 6,8±0,2. Wyjaławiać 15 minut w autoklawie w temperaturze 121°C, oziębić do 45–50°C i dodać sterylnego 1% roztworu telurytu potasu i 50 ml emulsji żółtka jaja kurzego.
7. 6. Podłoże do wykrywania Candida albicans
Pożywka Sabouard-dekstrozowa.
| Peptony (mięsny i kazeinowy) | 10,0 g |
| Dekstroza jednowodna | 40,0 g |
| Agar | 15,0 g |
| Woda destylowana | 1 000 ml |
Wyjaławiać 15 minut w autoklawie w temperaturze 121°C; pH po wyjałowieniu powinno wynosić 5,6±0,2.
Załącznik nr 2
METODY ANALIZ NIEZBĘDNYCH DO KONTROLI KOSMETYKU
l. Identyfikowanie i oznaczanie wolnego formaldehydu
1. Cel i zakres
Metoda opisuje identyfikowanie i dwie zasady oznaczania zależne od obecności lub nieobecności w próbce donorów formaldehydu. Metoda może służyć do badania wszystkich kosmetyków.
1. 1. Identyfikowanie
1. 2. Ogólne oznaczanie metodą kolorymetryczną z pentan-2,4-dionem
Tę metodę stosuje się, jeżeli używany jest formaldehyd sam lub z innymi środkami konserwującymi, które nie są donorami formaldehydu. W innym przypadku, gdy wynik przewyższa maksymalne dozwolone stężenie, należy zastosować poniższą metodę dla potwierdzenia wyniku.
1. 3. Oznaczenie w obecności donorów formaldehydu
W metodzie wspomnianej powyżej (1.2.) podczas otrzymywania pochodnych, donory formaldehydu ulegają rozczepieniu i prowadzi to do zawyżonych wyników (związany i spolimeryzowany formaldehyd). Konieczne jest wydzielenie wolnego formaldehydu metodą chromatografii cieczowej.
2. Definicja
Zawartość wolnego formaldehydu w próbce oznaczona według tej metody jest wyrażona w procentach masowych.
3. Identyfikowanie formaldehydu
3. 1. Zasada
Wolny i związany formaldehyd w środowisku kwasu siarkowego nadaje odczynnikowi Schiffa barwę różową lub fioletowo-różową
3.2. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej, woda musi być zdemineralizowana
3.2.1. Fuksyna
3.2.2. Siedmiowodny siarczan sodu
3.2.3. Stężony kwas solny (d = 1,19)
3.2.4. Kwas siarkowy, około 1 M
3.2.5. Odczynnik Schiffa:
100 mg fuksyny (3.2.1.) odważa się do zlewki i rozpuszcza w 75 ml wody w temperaturze 80°C. Po schłodzeniu dodaje się 2,5 g siarczynu sodu (3.2.2). Uzupełnia wodą do 100 ml.
Odczynnika można używać w ciągu dwóch tygodni.
3.3. Procedura
3.3.1. Zważyć 2 g próbki w 10 ml zlewce
3.3.2. Dodać dwie krople kwasu siarkowego (3.2.4) i 2 ml odczynnika Schiffa (3.2.5). Odczynnik ten podczas stosowania powinien być całkowicie bezbarwny. Wytrząsnąć i pozostawić do odstania na pięć minut.
3.3.3. Jeśli barwa różowa lub fioletowo-różowa pojawia się w ciągu pięciu minut, oznacza to, że formaldehyd jest obecny w stężeniu powyżej 0,01% i powinno się go oznaczyć metodą oznaczania wolnego i związanego formaldehydu (4), i, jeśli konieczne, według opisu postępowania podanego w punkcie 5.
4. Ogólne oznaczanie kolorymetryczne z pentan-2,4-dionem
4.1. Zasada
Formaldehyd reaguje z pentan-2,4-dionem w obecności octanu amonu, tworząc 3,5-diacetylo-1,4-dihydrotoluidynę. Jest ona ekstrahowana butan-1-olem i mierzy się absorbancję ekstraktu przy 410 nm.
4.2. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej, woda musi być zdemineralizowana.
4.2.1. Bezwodny octan amonu
4.2.2. Stężony kwas octowy, d420 = 1,05
4.2.3. Pentan-2,4-dion świeżo destylowany pod zmniejszonym ciśnieniem 25 mm Hg, w 25°C – nie powinien wykazywać żadnej absorbcji przy 410 nm.
4.2.4. Butan-1-ol
4.2.5. Kwas solny, 1 M
4.2.6. Kwas solny, około 0,1 M
4.2.7. Wodorotlenek sodu, 1 M
4.2.8. Świeżo przygotowany roztwór skrobi (1 g/50 ml wody)
4.2.9. Formaldehyd 37 do 40% (m/v)
4.2.10. Standardowy roztwór jodu, 0,05 M
4.2.11. Standardowy roztwór tiosiarczanu sodu, 0,1 M
4.2.12. Odczynnik pentan-2,4-dionu
W 1 000 ml kolbie miarowej rozpuścić: 150 g octanu amonu (4.2.1.), 2 ml pentan-2,4-dionu (4.2.3.), 3 ml kwasu octowego (4.2.2.). Uzupełnić wodą do 1 000 ml (pH roztworu około 6,4). Odczynnik ten powinien być świeżo przygotowany.
4.2.13. Odczynnik (4.2.12.) bez pentan-2,4-dionu
4.2.14. Standardowy bazowy roztwór formaldehydu
Nalać 5 g formaldehydu (4.2.9.) do 1 000 ml kolby miarowej i uzupełnić do 1 000 ml wodą. Oznaczyć stężenie tego roztworu następująco: pobrać 10,00 ml, dodać 25,00 ml standardowego roztworu jodu (4.2.10.) i 10,00 ml wodorotlenku sodu (4.2.7.). Pozostawić do odstania na pięć minut. Zakwasić 11,00 ml HCI (4.2.5.) i oznaczyć nadmiar jodu przez miareczkowanie standardowym roztworem tiosiarczanu sodu (4.2.11.), używając roztworu skrobi (4.2.8.) jako wskaźnika. 1 ml 0,05 M zużytego jodu (4.2.10.) odpowiada 1,5 mg formaldehydu.
4.2.15. Standardowy rozcieńczony roztwór formaldehydu
Rozcieńczyć standardowy bazowy roztwór formaldehydu wodą kolejno w stosunku 1/20 i następnie 1/100. 1 ml tego roztworu zawiera około 1 mg formaldehydu. Obliczyć dokładną zawartość.
4.3. Aparatura
4.3.1. Standardowe wyposażenie laboratoryjne
4.3.2. Sączek do rozdziału faz, bibuła Whatman (1PS lub równoważna)
4.3.3. Wirówka
4.3.4. Łaźnia wodna o temperaturze 60°C
4.3.5. Spektrofotometr
4.3.6. Szklane naczyńka pomiarowe o długości drogi optycznej 1 cm
4.4. Opis postępowania
4.4.1. Roztwór próbki
W 100 ml kolbie miarowej zważyć z dokładnością do 0,001 g ilość (w g) badanej próbki odpowiadającej spodziewanej zawartości formaldehydu około 150 µg. Uzupełnić wodą do 100 ml i wymieszać (roztwór S). (Sprawdzić, czy pH jest bliskie 6, jeśli nie – rozcieńczyć w roztworze kwasu solnego (4.2.6.)). Do 50 ml kolby stożkowej Erlenmayera dodawać: 10,00 ml roztworu S, 5,00 ml odczynnika pentan-2,4-dionu (4.2.12.), wodę zdemineralizowaną do końcowej objętości 30 ml.
4.4.2. Roztwór odniesienia
Możliwa interferencja powodowana przez barwę tła w badanej próbce jest eliminowana przez użycie takiego roztworu odniesienia. Do 50 ml kolby stożkowej Erlenmayera dodawać: 10,00 ml roztworu S, 5,00 ml odczynnika (14.2.13.), wodę zdemineralizowaną do końcowej objętości 30 ml.
4.4.3. Ślepa próba
Do 50 ml kolby stożkowej Erlenmayera dodawać: 5,00 ml roztworu odczynnika pentan-2,4-dionu (4.2.12,), wodę zdemineralizowaną do końcowej objętości 30 ml.
4.4.4. Oznaczanie
4.4.4.1. Wytrząsać mieszaniny z punktów 4.4.1., 4.4.2. i 4.4.3. Umieścić kolby stożkowe Erlenmayera w łaźni wodnej o temperaturze 60°C na dokładnie 10 minut. Pozostawić do schłodzenia na dwie minuty w łaźni wodnej z lodem.
4.4.4.2. Przenieść mieszaniny do 50 ml rozdzielaczy zawierających po 10 ml butan-1-olu (4.2.4.). Po-płukać każdą kolbę 3 do 5 ml wody. Wytrząsać energicznie mieszaninę przez dokładnie 30 sekund. Pozostawić do rozdzielenia.
4.4.4.3. Przesączyć fazę butan-1-olu do naczyńka pomiarowego (4.3.2.) przez sączek do rozdziału faz. Można również zastosować odwirowanie 3 000 obrotów w ciągu pięciu minut.
4.4.4.4. Zmierzyć absorbancję A1 ekstraktu roztworu z 4.4.1. przy 410 nm w porównaniu z ekstraktem 4.4.2. jako roztworem odniesienia.
4.4.4.5. Podobnie zmierzyć absorbancję A2 ekstraktu ślepej próby z punktu 4.4.3. w porównaniu z butan-1-olem jako roztworem odniesienia.
Uwaga. Wszystkie te czynności muszą być wykonane w ciągu 25 minut od chwili umieszczenia kolb stożkowych w łaźni wodnej o temperaturze 60°C.
4.4.5. Krzywa kalibracyjna
4.4.5.1. W 50 ml kolbie stożkowej Erlenmayera umieścić: 5,00 ml rozcieńczonego standardowego roztworu z punktu 4.2.15, 5,00 ml odczynnika pentan-2,4-dionu (4.2.12.), uzupełnić wodą zdemineralizowaną do końcowej objętości 30 ml.
4.4.5.2. Postępować dalej jak opisano w punkcie 4.4.4. i zmierzyć absorbancję w porównaniu z butan--1-olem (4.2.4.) jako roztworem odniesienia.
4.4.5.3. Powtórzyć operację z 10,15, 20 i 25 ml rozcieńczonego roztworu standardowego (4.2.15.)
4.4.5.4. Dla otrzymania wartości zerowej (odpowiadającej zabarwieniu odczynników) postępować jak w punkcie 4.4.4.5.
4.4.5.5. Wykreślić krzywą kalibracyjną po odjęciu wartości zerowej od każdej absorbancji oznaczonej w punktach 4.4.5.1. i 4.4.5.3. Prawo Beera obowiązuje do 30 µg formaldehydu.
4.5. Obliczenia
4.5.1. Odjąć A2 od A1 i odczytać z krzywej kalibracyjnej (4.4.5.5.) ilość C formaldehydu, w µg, w roztworze próbki (4.4.1.).
4.5.2. Obliczyć zawartość formaldehydu w próbce (% m/m) według następującego wzoru:
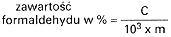
w którym:
m – masa próbki analitycznej w gramach.
4.6. Powtarzalność
Dla zawartości formaldehydu 0,2% różnica między wynikami dwu równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,005% przy oznaczaniu metodą kolorymetryczną z pentan-2,4-dionem. Jeśli oznaczenie wolnego formaldehydu prowadzi do wyników wyższych niż maksymalne stężenie ustalone:
a) pomiędzy 0,05% i 0,2% w wyrobie nieetykietowanym,
b) wyższe niż 0,2% w wyrobie zarówno etykietowanym, jak i nieetykietowanym, to znaczy, że należy postępować tak, jak podano w punkcie 5 poniżej.
5. Oznaczanie formaldehydu w obecności donorów formaldehydu
5.1. Zasada
Wydzielony formaldehyd przeprowadzany jest w żółtą pochodną lutydynową w reakcji z pentan-2,4-dionem w reaktorze po kolumnie rozdzielającej i otrzymana pochodna jest oznaczana przez pomiar absorbancji przy 420 nm.
5.2. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej, a woda musi być zdemineralizowana.
5.2.1. Woda czystości odpowiedniej dla HPLC lub woda równoważnej jakości
5.2.2. Bezwodny octan amonu
5.2.3. Stężony kwas octowy
5.2.4. Pentan-2,4-dion (przechowywany w 4°C)
5.2.5. Bezwodny fosforan dwusodowy
5.2.6. 85% kwas ortofosforowy (d = 1,7)
5.2.7. Metanol o czystości dla HPLC
5.2.8. Dichlorometan
5.2.9. Formaldehyd 37 do 40% m/v
5.2.10. Wodorotlenek sodu, 1 M
5.2.11. Kwas solny, 1 M
5.2.12. Kwas solny, 0,002 M
5.2.13. Roztwór skrobi świeżo przygotowany według Farmakopei Europejskiej (p. 4.2.8.)
5.2.14. Standardowy roztwór jodu, 0,005 m.
5.2.15. Standardowy roztwór tiosiarczanu sodu, 0,1 m.
5.2.16. Faza ruchoma
Wodny roztwór fosforanu dwusodowego (5.2.5.), 0,006 M nastawiony na pH 2,1 kwasem ortfosforowym (5.2.6.)
5.2.17. Odczynnik dla mediów schodzących z kolumny („po kolumnie”)
W 1 000 ml kolbie miarowej rozpuścić: 62,5 g octanu amonu (5.2.2.), 7,5 g kwasu octowego (5.2.3.), 5 ml pentan-2,4-dionu (5.2.4). Uzupełnić wodą do 1 000 ml (5.2.1.). Przechowywać odczynnik bez dostępu światła. Okres przechowywania: maksymalnie trzy dni w temperaturze 25°C. Nie powinny zachodzić żadne zmiany w zabarwieniu odczynnika.
5.2.18. Standardowy bazowy roztwór formaldehydu
Wlać 10 g formaldehydu (5.2.9.) do 1 000 ml kolby miarowej i uzupełnić wodą do 1 000 ml. Oznaczyć stężenie tego roztworu następująco. Pobrać 5,00 ml, dodać 25,00 ml standardowego roztworu jodu (5.2.14.) i 10,00 ml roztworu wodorotlenku sodowego (5.2.10.). Pozostawić do odstania na 5 minut. Zakwasić 11,00 ml HCI (5.2.11.) i miareczkować nadmiar standardowego roztworu jodu standardowym roztworem tiosiarczanu sodu (5.2.15.), używając roztworu skrobi (5.2.13.) jako wskaźnika. 1 ml roztworu jodu (5.2.14.) jest równoważnikiem 1,5 mg formaldehydu.
5.2.19. Standardowy rozcieńczony roztwór formaldehydu
Rozcieńczyć bazowy roztwór do 1/100 jego początkowego stężenia w fazie ruchomej (5.2.16.). 1 ml tego roztworu zawiera około 37 mg formaldehydu. Obliczyć jego dokładną zawartość.
5.3. Aparatura
5.3.1. Standardowe wyposażenie laboratoryjne
5.3.2. Pompa HPLC o podawaniu niepulsacyjnym
5.3.3. Niskociśnieniowa pompa o podawaniu niepulsacyjnym dla odczynnika (lub druga pompa HPLC)
5.3.4. Zawór zastrzykowy do wprowadzania próbki z pętlą 10 µl
5.3.5. Reaktor mieszaniny schodzącej z kolumny („po kolumnie”) z następującymi elementami:
a) jedna 1-litrowa kolba trójszyjna,
b) jeden płaszcz grzewczy dla kolby 1 litrowej,
c) dwie kolumny Vigreux o minimum 10 półkach z dwoma płaszczami chłodzonymi powietrzem,
d) rurka ze stali kwasoodpornej (dla wymiennika ciepła) 1,6 mm, wewnętrzna średnica 0,23 mm, długość 400 mm,
e) rurka teflonowa 1,6 mm, wewnętrzna średnica 0,30 mm, długość 5 m (splot francuski) (zob. zał. 1),
f) jeden łącznik T-kształtny bez martwej objętości (Valco lub równoważny),
g) trzy łączniki bez martwej objętości lub jeden moduł „po kolumnie” Applied Biosystems PCRS 520 lub równoważny uzupełniony o 1 ml reaktor
5.3.6. Filtr membranowy o średnicy porów 0,45 µm.
5.3.7. Ładunek filtru SEP-PAK lub równoważny
5.3.8. Kolumny przygotowane fabrycznie:
a) Bischoff hypersil RP (typ NC, odnośnik C 25, 46 1805) (5 µm, długość = 250 mm, średnica wewnętrzna = 4,6 mm),
b) lub Dupont, Zorbax ODS (5 µm, długość 250 mm, średnica wewnętrzna = 4,6 mm),
c) lub Phase SEP, spherisorb ODS (5 µm, długość 250 mm, średnica wewnętrzna = 4,6 mm).
5.3. 9. Kolumna wstępna
Bischoff K1. hypersil RP 18 (odnośnik K1G 6301 1805) (5 µm, długość 10 mm, lub równoważny).
5.3.10. Kolumna i kolumna wstępna są połączone układem Ecotube (odnośnik A 150 20508 Bischoff) lub równoważnym.
5.3.11. Aparaturę (5.3.5.) złożyć tak, jak pokazano na schemacie blokowym (rys. 6).
Połączenia te po zastrzykowym zaworze wprowadzającym muszą być możliwie najkrótsze. W tym przypadku rurka ze stali kwasoodpornej między wylotem reaktora i wlotem detektora jest przeznaczona do schłodzenia mieszaniny przed detekcją i temperatura w detektorze jest nieznana, lecz stała.
5.3.12. Detektor UV i promieniowania widzialnego
5.3.13. Rejestrator
5.3.14. Wirówka
5.3.15. Łaźnia ultradźwiękowa (płuczka wibracyjna)
5.3.16. Mieszadło wibracyjne (Vortex lub równoważne)
5.4. Procedura
5.4.1. Krzywa kalibracyjna
Tworzy się ją przez wykreślenie wysokości pików jako funkcji stężenia rozcieńczonego standardowego roztworu formaldehydu. Przygotować standardowe roztwory przez rozcieńczenie roztworu odniesienia formaldehydu (5.2.19.) fazą ruchomą (5.2.16.);
a) 1,00 ml roztworu (5.2.19.) rozcieńczone do 20,00 ml (około 185 µg/100 ml),
b) 2,00 ml roztworu (5.2.19.) rozcieńczone do 20,00 ml (około 370 µg/100 ml),
c) 5,00 ml roztworu (5.2.19.) rozcieńczone do 25,00 ml (około 740 µg/100 ml),
d) 5,00 ml roztworu (5.2.19.) rozcieńczone do 20,00 ml (około 925 µg/100 ml).
Te roztwory standardowe mogą być przechowywane w ciągu jednej godziny w temperaturze laboratorium i muszą być świeżo przygotowane. Krzywa kalibracyjna ma właściwy przebieg liniowy dla stężeń pomiędzy 1,00 i 15,00 µg/rnl.
5.4.2. Przygotowanie próbek
5.4.2.1. Emulsje (kremy, bazy do makijażu, tusze do oczu)
W 100 ml kolbie z korkiem zważyć z dokładnością do 0,001 g ilość próbki analitycznej (m. g) odpowiadającą przewidywanej ilości 100 µg formaldehydu. Dodać 20,00 ml dichlorometanu (5.2.8.) i 20,00 ml kwasu solnego (5.2.12.) dokładnie odmierzonych. Mieszać mieszadłem wibracyjnym (5.3.16.) i w płuczce wibracyjnej (5.3.15.). Rozdzielić dwie fazy przez wirowanie 3.000 gn w ciągu 2 minut). W międzyczasie przepłukać ładunek filtru (5.3.7.) 2 ml metanolu (5.2.7.), następnie kondycjonować go 5 ml wody (5.2.1.). Przepuścić 4 ml fazy wodnej ekstraktu przez kondycjonowany ładunek filtru, odrzucić pierwsze 2 ml i zachować następną frakcję.
5.4.2.2. Płyny, szampony
W 100 ml kolbie z korkiem zważyć z dokładnością do 0,001 g próbkę analityczną (m. g) w ilości odpowiadającej przewidywanej ilości około 500 µg formaldehydu. Uzupełnić do 100 ml fazą ruchomą (5.2.16.). Przesączyć roztwór przez filtr (5.3.6.) i wstrzyknąć lub przepuścić przez ładunek filtru (5.3.7.) kondycjonowany tak jak poprzednio (5.4.2.1.). Przed ponownym kondycjonowaniem układu należy przepuścić przez filtr wodę, aby uniknąć rekrystalizacji. Wszystkie roztwory muszą być wstrzykiwane bezpośrednio po przygotowaniu.
5.4.3. Warunki chromatograficzne:
a) szybkość przepływu fazy ruchomej: 1 ml/min,
b) szybkość przepływu odczynnika: 0,5 ml/min,
c) całkowita szybkość przepływu przy wylocie z detektora: 1,5 ml/min,
d) wstrzykiwana objętość: 10 µl,
e) temperatura eluowania: w przypadku trudności w rozdziale należy zanurzyć kolumnę w łaźni lodowo-wodnej i poczekać 15-20 minut na ustabilizowanie się temperatury,
f) temperatura reakcji po kolumnie: 100°C,
g) detekcja: 420 nm
Cały układ chromatograficzny łącznie z reaktorem po kolumnie musi być przepłukany wodą po użyciu (5.2.1.). Jeśli układ nie jest używany dłużej niż w ciągu dwóch dni, to po płukaniu wodą należy układ przepłukać metanolem (5.2.7.). Przed ponownym kondycjonowaniem należy układ przepłukać wodą, aby uniknąć krystalizacji.
5.5. Obliczenia
Emulsje: (5.4.2.1.):
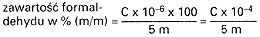
Płyny, szampony:
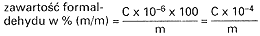
gdzie:
m – masa analizowanej próbki w g (5.4.2.1),
C – stężenie formaldehydu w mg/100 ml odczytane z krzywej kalibracyjnej (5.4.1.).
5.6. Powtarzalność.
Dla zawartości formaldehydu 0,05% różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,001%.
Dla zawartości formaldehydu 0,2% różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,005 %.
INSTRUKCJA WYKONANIA „SPLOTU FRANCUSKIEGO”
Wymagane przybory
a) Jedna drewniana szpula o średnicy zewnętrznej 5 cm z otworem o średnicy 1,5 cm przechodzącym przez środek. Umieścić cztery stalowe gwoździe (jak pokazano na rys, 1 i 2). Odległość między dwoma gwoździami musi wynosić 1,8 cm, a gwoździa od otworu 0,5 cm (rys. 2).
b) Jedna sztywna igła ( w formie rozwidlonego haczyka) dla zaczepienia rurki teflonowej.
c) Rurka teflonowa długości 5 m o średnicy zewnętrznej 1,6 mm i średnicy wewnętrznej 0,3 mm.
PROCEDURA
Dla rozpoczęcia „splotu francuskiego" należy przeprowadzić rurkę teflonowa od góry do dołu przez otwór szpuli (pozostawiając około 10 cm rurki wystającej z dolnego końca szpuli, aby umożliwić tworzenie łańcucha podczas procesu wykonywania splotu); następnie nawinąć rurkę na gwoździe, jak pokazano na rys.3. Góra i dół splotu francuskiego będą chronione metalowymi pierścieniami i śrubami, należy uważać, aby nie zgnieść rurki teflonowej podczas zaciskania splotu. Nawinąć rurkę na około każdego gwoździa po raz drugi i zrobić „oczka" następująco: podnieść dolną rurkę nad górną z pomocą haczyka (patrz rys.4). Powtórzyć te czynności z każdym gwoździem w kolejności (1, 2, 3, 4 w kierunku przeciwnym do kierunku wskazówek zegara) do momentu uzyskania 5 m lub wymaganej długości splotu.
Pozostawić około 10 cm rurki na zakończenie łańcucha. Przewlec rurkę przez każdą z czterech pętli i przeciągnąć delikatnie w celu zamknięcia końca łańcucha.
Rys. 1. Schematyczny rysunek szpuli
Rys. 2. Umiejscowienie gwoździ na szpuli
Rys. 3. Pierwsza rurka
Rys. 4. Druga rurka. Aby wykonać „oczko", podnieść dolną rurkę (linia ciągła) powyżej drugiej rurki – linia przerywana

Rys. 5
Rys. 6:
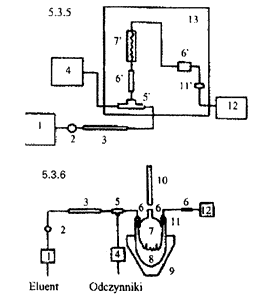
1. Pompa HPLC
2. Zawór zastrzykowy do wprowadzania próbki
3. Kolumna z kolumną wstępną
4. Pompa dla odczynnika
5. Łącznik T(vortex)
5'. Łącznik T-kształtny bez martwej objętości
6-6'. Połączenie bez martwej objętości
7. Splot francuski
7'. Reaktor
8. Trójszyjna kolba z wrzącą wodą
9. Płaszcz grzewczy dla kolby
10. Chłodnica
11. Rurka wymiennika ciepła ze stali kwasoodpornej
12. Detektor w zakresie promieniowania DV i widzialnego
13. Zestaw po kolumnie PCRS-520
II. Identyfikowanie i półilościowe oznaczanie wybranych barwników utleniających w farbach do włosów
1. Cel i zakres
Metoda jest odpowiednia do identyfikowania i półilościowego oznaczania następujących substancji w farbach do włosów w postaci kremu lub płynu:
| Substancje | Symbol |
| Fenylenodiaminy |
|
| o-fenylenodiamina | (OPD) |
| m-fenylenodiamina | (MPD) |
| p-fenylenodiamina (Aneks V Dyrektywy Kosmetycznej) | (PPD) |
| Metylofenylenodiaminy |
|
| 4-metylo-1,2-fenylenodiamina (3,4-diaminotoluen) | (OTD) |
| 4-metylo-1,3-fenylenodiamina (2,4-diaminotoluen) | (MTD) |
| 2-metylo-1,4-fenylenodiamina (2,5-diaminotoluen) | (PTD) |
| Diaminofenole |
|
| 2,4-diaminofenol | (DAP) |
| Hydrochinon |
|
| 1,4-benzenodiol | (H) |
| a-naftol | (a-N) |
| Pirogallol |
|
| 1,2,3-trihydroksybenzen | (P) |
| Rezorcyna |
|
| 1,3-dihydroksybenzen | R |
2. Zasada
Barwniki utleniające ekstrahuje się z farb w postaci kremu lub płynu 96% etanolem przy pH 10 i identyfikuje metodą chromatografii cienkowarstwowej, jedno- i dwukierunkowej. Dla półilościowego oznaczania tych substancji chromatogramy próbek otrzymane z pomocą czterech układów rozwijających są porównywalne z chromatogramami substancji odniesienia otrzymanymi w tym samym czasie i w warunkach tak podobnych, jak jest to możliwe.
3. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej
3.1. Bezwodny etanol
3.2. Aceton
3.3. Etanol, 96% (v/v)
3.4. Roztwór amoniaku, 25% (d420 = 0,91)
3.5. Kwas L(+)askorbinowy
3.6. Chloroform
3.7. Cykloheksan
3.8. Azot techniczny
3.9. Toluen
3.10. Benzen
3.11. n-Butanol
3.12. Butan-2-ol
3.13. Kwas podfosforawy, roztwór 50% (v/v)
3.14. Odczynniki do diazowania, jeden z dwóch do wyboru:
a) chlorobenzenosulfonian 3-nitrobenzenodiazoniowy (stabilizowany w postaci soli), taki jak odczynnik Red 2 NJ Francolor,
b) 2-naftalenosulfonian 2-chloro-4-nitrobenzenodiazoniowy (stabilizowany w postaci soli ), taki jak odczynnik NNCD Reagent Nr 74150 Fluka lub równoważny.
3.15. Azotan srebra
3.16. Aldehyd p.-dimetyloaminobenzoesowy
3.17. 2,5-Dimetylofenol
3.18. Chlorek żelaza sześciowodny
3.19. Kwas solny, roztwór 10% m/v
3.20. Substancje odniesienia.
Substancje odniesienia przedstawiono w ustępie 1 „Cel i zakres". W przypadku związków aminowych, substancją odniesienia mogą być chlorowodorek (mono- lub di-) lub wolna zasada.
3.21. Roztwory odniesienia 0,5% (m/v).
Przygotowuje się 0,5% (m/v) roztwory wszystkich substancji odniesienia podanych w punkcie 3.20. Odważyć 50 mg ±1 mg substancji odniesienia w 10 ml kolbie miarowej. Dodać 5 ml 96% etanolu (3.3.), 250 mg kwasu askorbinowego (3.5) i wymieszać. Zalkalizować, dodając roztwór amoniaku (3.4.) do uzyskania wyraźnego odczynu o pH 10 (zbadać papierkiem wskaźnikowym). Uzupełnić 96% etanolem (3.3.) do 10 ml i wymieszać. Roztwory można przechowywać w ciągu tygodnia w chłodnym miejscu bez dostępu światła. W pewnych przypadkach, po dodaniu kwasu askorbinowego i amoniaku, może wytrącać się osad. Należy wówczas pozwolić na jego osadzenie przed zastosowaniem.
3.22. Rozpuszczalniki rozwijające.
3.22.1. Aceton-chloroform-toluen (35:25:40 obj.)
3.22.2. Chloform-cykloheksan-etanol absolutny – 25% amoniak (80:10:10 :1obj.)
3.22.3. Benzen-butan-2-ol – woda (50:25:25 obj.)
Wytrząsać dobrze i stosować górną fazę po rozdzieleniu w temperaturze pokojowej (20 do 250C).
3.22.4. n-Butanol-chloroform – odczynnik M (7:70:23 obj.)
Rozdzielać starannie w temperaturze pokojowej (20 do 250C i stosować dolną fazę.
Przygotowanie odczynnika M:
| 25% (v/v) roztwór amoniaku | 24 objętości |
| 50% kwas podfosforawy (3.13.) | 1 objętość |
| woda | 75 objętości |
Rozpuszczalniki rozwijające zawierające amoniak należy dobrze wytrząsać bezpośrednio przed użyciem.
3.23. Rozpylane roztwory wywołujące
3.23.1. Odczynnik do diazowania
Przygotować 5% (m/v) wodny roztwór wybranego odczynnika (3.14). Roztwór ten należy przygotować bezpośrednio przed użyciem.
3.23.2. Odczynnik Ehrlicha
Rozpuścić 2 g aldehydu p.-dimetyloaminobenzoesowego (3.16.). w 100 ml 10% (m/v) wodnego roztworu kwasu solnego (3.19.).
3.23.3. 2,5-dimetylofenol – sześciowodny chlorek żelaza
Roztwór 1: Rozpuścić 1 g dimetylofenolu (3.17.) w 100 ml 96% etanolu (3.3.)
Roztwór 2: Rozpuścić 4 g sześciowodnego chlorku żelaza (3.18.) w 100 ml 96% etanolu (3.3.)
Do wywoływania należy te roztwory rozpylać oddzielnie, najpierw roztwór 1, następnie roztwór 2.
3.23.4. Amoniakalny azotan srebra
Do 5% (m/v) wodnego roztworu azotanu srebra (3.15.) dodaje się 25% amoniak aż do rozpuszczenia osadu. Odczynnik ten musi być przygotowany bezpośrednio przed użyciem. Nie należy go przechowywać.
4. Aparatura
4.1. Standardowe wyposażenie laboratoryjne do chromatografii cienkowarstwowej.
4.1.1. Osłona z tworzywa lub szklana tak skonstruowana, aby azot mógł opływać płytkę chromatograficzną podczas nanoszenia kropli próbek i suszenia. Ostrożność ta jest konieczna ze względu na podatność pewnych barwników na utlenianie.
4.1.2. Mikrostrzykawka 10 µl, kalibrowana z podziałkami 0,2 µl, z okrągło zakończoną igłą lub lepiej 50 µl wielokrotny dozownik, zmontowany na statywie śrubowym w taki sposób, że płytka może być utrzymywana w atmosferze azotu.
4.1.3. Cienkowarstwowe płytki krzemionkowe gotowe do stosowania, o grubości 0,25 mm, o wymiarach 20 x 20 cm (firmy Macherey and Nagel, Silica G-HR, które mają podłoże tworzywowe lub równoważne).
4.2. Wirówka 4 000 obrotów/minutę
4.3. Probówki do wirówki, 10 ml z korkiem gwintowanym pokryte PTFE lub równoważne.
5. Procedura
5.1 Przygotowanie próbek
Odrzucać pierwsze 2 lub 3 cm kremu wyciśniętego z tuby. Umieścić następujące materiały w probówce wirówki (4.3.) uprzednio przepłukanej azotem; 300 mg kwasu askrobinowego, z 3 g kremu lub 3 g homogenizowanego płynu. Dodać kroplami 25% amoniak (3.4.) do uzyskania pH 10.
Uzupełnić 96% etanolem (3.3.) do 10 ml. Homogenizować w atmosferze azotu (3.8.), zamknąć i następnie wirować przy 4 000 obrotów/minutę w ciągu 10 minut. Do analizy stosować ciecz znajdującą się na górze.
5.2. Chromatografia
5.2.1. Nanoszenie kropli na płytki
W atmosferze azotu (3.8.) nanieść na płytkę chromatograficzną (4.1.3.) po 1 µl wszystkich opisanych powyżej roztworów odniesienia w dziewięciu punktach umieszczonych w odległości po około 1,5 cm wzdłuż linii znajdującej się średnio 1,5 cm od krawędzi płytki. Naniesienia roztworów odniesienia należy rozmieścić następująco:
| 1 | 2 | 3 | 4 | 5 |
| R | P | H | PPD | DAP |
| MTD | a-N |
|
|
|
|
|
|
|
|
|
| 6 | 7 | 8 | 9 |
|
| PTD | OPD | OTD | MPD |
|
Dodatkowo nanieść w punktach 10 i 11 odpowiednio po 2 µi próbek badanych roztworów otrzymanych zgodnie z opisem w punkcie 5.1. Utrzymywać płytkę w atmosferze azotu aż do chwili, kiedy jest podana rozdziałowi chromatograficznemu.
5.2.2. Rozwijanie
Umieścić płytkę w komorze uprzednio przepłukanej azotem (3.8.) wysyconej jednym z czterech rozpuszczalników (3.22.) i pozostawić do rozwijania w temperaturze pokojowej (20 do 250C, w ciemności do chwili, kiedy czoło rozpuszczalnika osiągnie wysokość około 15 cm od linii bazowej.
Wyjąć płytkę z komory i suszyć w atmosferze azotu (3.8.) w temperaturze pokojowej.
5.2.3. Spryskiwanie
Spryskać płytkę bezzwłocznie jednym z czterech rozpuszczalników wymienionych w punkcie 3.23.
5.2.4. Identyfikowanie
Porównać wartości Rf i barwy otrzymanych plam z tymi otrzymanymi dla substancji odniesienia poddanych chromatograficznemu rozdziałowi. Tab. l podaje przykłady wartości Rf i barwy dla każdej substancji zależnie od stosowanych rozpuszczalników i środków wywołujących. Potwierdzenie wątpliwego identyfikowania można czasem uzyskać metodą wzorca wewnętrznego, dodając roztwór odpowiedniej substancji odniesienia do ekstraktu próbki.
5.2.5. Ocena półilościowa
Porównać wizualnie intensywność plam dla każdej substancji identyfikowanej w punkcie 5.2.4. z odpowiednim zakresem stężeń substancji odniesienia. Jeśli stężenie jednej lub więcej substancji znalezionych w próbce jest zbyt wysokie, rozcieńczyć ekstrakt próbki i powtórzyć pomiar.
Tab. l. Wartości Rf i barwy otrzymane bezpośrednio po spryskaniu
| Substancja odniesienia (3.20) | Rozpuszczalniki rozwijające | Rozpylane roztwory rozwijające | ||||||
| wartości RF | otrzymane barwy | |||||||
| (3.22.1.) | (3.22.2.) | (3.22.3.) | (3.22.4.) | Diazowy (3.23.1.) | Ehrlich (3.23.2.) | Dimetylofenol (3.23.3.) | AgNO3 (3.24.4.) | |
| OPD | 0,62 | 0,60 | 0,30 | 0,57 | bladobrązowa | – | – | bladobrązowa |
| MPD | 0,40 | 0,60 | 0,47 | 0,48 | fioletowo-brązowa(*) | żółta | bladobrązowa | bladobrązowa |
| PPD | 0,20 | 0,50 | 0,30 | 0,48 | brązowa | jasnoczerwona (*) | fioletowa (*) | szara |
| OTD | 0,60 | 0,60 | 0,53 | 0,60 | brązowa (*) | bladopomarańczowa | bladobrązowa | szarawobrązowa |
| MPD | 0,40 | 0,67 | 0,45 | 0,60 | czerwonawo--brązowa (*) | żółta | brązowa | czarna |
| PTD | 0,33 | 0,65 | 0,37 | 0,70 | brązowa | pomarańczowa | fioletowa (*) | szara |
| DAP | 0,07 | – | 0 | 0,05 | brązowa (*) | pomarańczowa | fioletowa | brązowa |
| H | 0,50 | 0,35 | 0,80 | 0,20 | – | pomarańczowa | fioletowa | czarna (*) |
| a-N | 0,90 | 0,80 | 0,90 | 0,75 | pomarańczowo-brązowa | – | fioletowa (*) | czarna |
| P | 0,37 | – | 0,67 | 0,05 | brązowa | bardzo bladofioletowa | bardzo bladobrązowa | brązowa (*) |
| R | 0,50 | 0,37 | 0,80 | 0,17 | pomarańczowa(*) | bladofioletowa | bardzo bladobrązowa | bladobrązowa |
Uwaga
OPD jest tylko słabo widoczny; należy użyć rozpuszczalnika (3.22.3), aby rozdzielić go od OTD.
(*) Wskazuje najlepszą wywołaną barwę.
6. Badanie metodą dwukierunkowej chromatografii cienkowarstwowej
Procedura chromatografii dwukierunkowej wymaga użycia dodatkowych substancji standardowych i odczynników
6.1. Dodatkowe roztwory i substancje odniesienia
6.1.1. β-naftol (b-N)
6.1.2. 2-aminofenol (OAP)
6.1.3. 3-aminofenol (MAP)
6.1.4. 4-aminofenol (PAP)
6.1.5. 2-nitro-1,4-fenylenodiamina (2-NPPD)
6.1.6. 4-nitro-1,2-fenylenodiamina (4-NOPD)
Przygotować 0,5% (m/v) roztworów wszystkich dodatkowych substancji odniesienia jak opisano w punkcie 3.21.
6.2. Dodatkowy rozpuszczalnik rozwijający
6.2.1. Octan etylu-cykloheksan-25% roztwór amoniaku (65:30:0,5 obj.)
6.3. Dodatkowy układ wywołujący
Umieścić szklane naczynie w komorze do chromatografii cienkowarstwowej, dodać około 2 g kryształów jodu i zamknąć komorę odpowiednią pokrywką.
6.4. Chromatografia
6.4.1. Narysować dwie linie (rys. 1) na powierzchni adsorbenta płytki cienkowarstwowej (4.1.3.).
6.4.2. W atmosferze azotu (4.1.1.) nanieść 1 do 4 µl ekstraktu (5.1.) w punkcie bazowym 1 (rys. 1), który znajduje się w odległości 2 cm od obu krawędzi. Ilość naniesionego ekstraktu zależy od intensywności plam na chromatogramach (5.2).
6.4.3. Między punktami 2 i 3 rozdzielać (rys. 1) barwniki utleniające zidentyfikowane lub uważane za zidentyfikowane przez rozdział chromatograficzny (5.2.) (odległość między punktami wynosi 1,5 cm). Nanosić po 2 µl wszystkich roztworów odniesienia, z wyjątkiem DAP, który trzeba nanieść w ilości 6 µl. Działania prowadzić w atmosferze azotu (6.4.2.).
6.4.4. Powtórzyć operację z punktu 6.4.3. w punktach bazowych 4 i 5 (rys. 1) i utrzymywać płytkę w atmosferze azotu aż do chwili rozpoczęcia rozdziału chromatograficznego (odległość między punktami wynosi 1,5 cm).
6.4.5. Przepłukać komorę chromatograficzną azotem (3.8.) i umieścić w niej odpowiednią ilość rozpuszczalnika rozwijającego (3.22.2). Umieścić płytkę (6.4.4.) w komorze i rozwijać ją w ciemności w pierwszym kierunku elucji (rys. 1). Eluować do chwili, kiedy czoło rozpuszczalnika osiągnie linię zaznaczoną na płytce (około 13 cm).
6.4.6. Wyjąć płytkę z komory i umieścić ją w komorze chromatograficznej przepłukanej uprzednio azotem przez co najmniej 60 minut, aby odparować rozpuszczalnik eluujący.
6.4.7. Za pomocą kalibrowanej probówki wprowadzić odpowiednią ilość rozpuszczalnika eluującego (6.2.) do komory przepłukanej azotem (3.8.), umieścić płytkę obróconą o 900 w komorze (6.4.6.), wprowadzić rozdział chromatograficzny w drugim kierunku (również w ciemności) do chwili, kiedy czoło rozpuszczalnika osiągnie linię narysowaną na powierzchni adsorbenta. Wyjąć płytkę z komory i odparować rozpuszczalnik eluujący na powietrzu.
6.4.8. Umieścić płytkę w komorze chromatograficznej wysyconej parami jodu (6.3.) na 10 minut i interpretować dwukierunkowy chromatogram korzystając z wartości Rf i walorów barwy substancji odniesienia rozdzielanych chromatograficznie w tym samym czasie (tab. II stanowi przewodnik po wartości Rf i walorach barw).
Uwaga. Dla otrzymania maksymalnego wybarwienia plam należy pozostawić chromatogram wystawiony na działanie powietrza w ciągu pół godziny po rozwinięciu.
6.4.9. Obecność barwników utleniających stwierdzoną w punkcie 6.4.8. można wyraźnie potwierdzić przez powtórzenie operacji opisanych w punktach 6.4.1. do 6.4.8. i dodanie w 1 punkcie bazowym do ilości ekstraktu podanej w punkcie 6.4.2. 1 µl substancji odniesienia zidentyfikowanej w punkcie 6.4.8. Jeśli nie zostanie znaleziona dodatkowa plama w porównianiu z chromatogramem otrzymanym według punktu 6.4.8., interpretacja chromatogramu z punktu 6.4.8. jest prawidłowa.
Tab. II. Barwa substancji odniesienia po procesie chromatografii i wywołaniu parami jodu
| Substancja odniesienia | Barwa po wywołaniu parami jodu |
| R | beżowa |
| P | brązowa |
| alfa-N | fioletowa |
| beta-N | bladobrązowa |
| H | fioletowo-brązowa |
| MPD | żółtawobrązowa |
| PPD | fioletowo-brązowa |
| MTD | ciemnobrązowa |
| PTD | żółtawobrązowa |
| DAP | ciemnobrązowa |
| OAP | pomarańczowa |
| MAP | żółtawobrązowa |
| PAP | fioletowo-brązowa |
| 2-NPPD | brązowa |
| 4-NOPD | pomarańczowa |
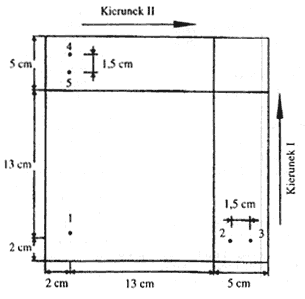
Rys. 1
III. Identyfikacja i oznaczanie kwasu szczawiowego i jego soli alkalicznych w środkach do pielęgnacji włosów
1. Cel i zakres metody
Opisana poniżej metoda jest odpowiednia do identyfikacji i oznaczania kwasu szczawiowego i jego soli alkalicznych w środkach do pielęgnacji włosów. Może być ona stosowana dla bezbarwnych wodnych/alkoholowych roztworów i płynów, które zawierają około 5% kwasu szczawiowego lub równoważną ilość alkalicznych szczawianów.
2. Definicja
Zawartość kwasu szczawiowego lub jego soli alkalicznych oznaczona według tej metody jest wyrażona jako zawartość w procentach masowych m/m wolnego kwasu szczawiowego w próbce.
3. Zasady
Po usunięciu wszystkich obecnych anionowych środków powierzchniowo czynnych chlorowodorkiem p-toluidyny, kwas szczawiowy i/lub szczawiany wytrącane są jako szczawian wapnia, po czym roztwór jest przesączany. Osad jest rozpuszczany w kwasie siarkowym i miareczkowany roztworem nadmanganianu potasu.
4. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej
4.1. 5% (m/m) roztwór octanu amonu,
4.2. 10% (m/m) roztwór chlorku wapnia,
4.3. 95% (v/v) etanol,
4.4. czterochlorek węgla,
4.5. eter dietylowy,
4.6. 6–8% (m/m) roztwór dichlorowodorku p-toluidyny,
4.7. 0.1 N roztwór nadmanganianu potasu,
4.8. 20% (m/m) kwas siarkowy,
4.9. 10% (m/m) kwas chlorowodorowy,
4.10. trójwodny octan sodu,
4.11. kwas octowy lodowaty,
4.12. kwas siarkowy (1:1),
4.13. nasycony roztwór wodorotlenku baru.
5. Aparatura
5.1. rozdzielacze 500 ml
5.2. zlewki 50 i 600 ml
5.3. tygle z filtrem szklanym G-4
5.4. cylindry miarowe 25 i 100 ml
5.5. pipety 10 ml
5.6. kolby ssawkowe 500 ml
5.7. próżniowa pompa wodna (smoczek parowy)
5.8. termometr o zakresie skali 0–1000C
5.9. mieszadło magnetyczne z elementem grzewczym
5.10. pręty magnetyczne pokryte teflonem
5.11. biureta 25 ml
5.12. kolby stożkowe 250 ml.
6. Procedura
6.1. Odważyć 6–7 g próbki do zlewki o pojemności 50 ml, doprowadzić do pH 3 rozcieńczonym kwasem chlorowodorowym (4.9) i myć w rozdzielaczu 100 ml wody destylowanej. Dodawać stopniowo 25 ml etanolu (4.3), 25 ml roztworu dichlorowodorku p-toluidyny (4.6) i 25 do 30 ml czterochlorku węgla (4.4) i energicznie wytrząsać mieszaninę.
6.2. Po rozdzieleniu faz usunąć dolną (organiczną) fazę, powtórzyć ekstrakcję, używając reagentów podanych w punkcie 6.1. i znowu usunąć fazę organiczną.
6.3. Przelać wodny roztwór do zlewki o pojemności 600 ml i usunąć obecny jeszcze czterochlorek węgla przez ogrzewanie roztworu do wrzenia.
6.4. Dodać 50 ml roztworu octanu amonu (4.1), doprowadzić roztwór do wrzenia (5.9) i wmieszać 10 ml gorącego roztworu chlorku wapnia (4.2) do wrzącego roztworu; pozwolić wytrącić się osadowi.
6.5. Sprawdzić, czy wytrącenie osadu jest całkowite, dodając kilka kropli roztworu chlorku wapnia (4.2), pozostawić do schłodzenia do temperatury pokojowej i następnie wmieszać do 200 ml etanolu (4.3); (5.10) pozostawić do odstania na 30 minut.
6.6. Przesączyć ciecz przez tygiel z filtrem szklanym (5.3), przenieść osad z małą ilością gorącej wody (50 do 600C) do tygla z filtrem szklanym i myć osad zimną wodą.
6.7. Przemyć osad pięć razy małą ilością etanolu (4.3) i potem pięć razy małą ilością eteru etylowego (4.5) i rozpuścić osad w 50 ml gorącego kwasu siarkowego (4.8) przez przeciąganie tego ostatniego przez szklany filtr tygla pod zmniejszonym ciśnieniem.
6.8. Przenieść roztwór bez strat do kolby stożkowej (5.1.1) i miareczkować roztworem nadmanganianu potasu (4.7) aż do wystąpienia jasnoróżowego zabarwienia.
7. Obliczenia
Zawartość kwasu szczawiowego w próbce wyrażoną jako procent masowy oblicza się z wzoru:
| % kwasu szczawiowego = | A x 4.50179 x 100 |
| E x 1000 |
w którym:
A oznacza zużyty 0.1 N roztwór nadmanganianu potasu zmierzony zgodnie z p. 6.8,
E oznacza masę próbki analitycznej wyrażoną w gramach (6.1),
4.50179 oznacza współczynnik przeliczeniowy kwasu szczawiowego.
8. Powtarzalność
Dla zawartości kwasu szczawiowego wynoszącej około 5% różnica pomiędzy wynikami z dwóch równolegle prowadzonych analiz na tej samej próbce nie powinna przekraczać wartości bezwzględnej 0,15%.
9. Identyfikowanie
9.1. Zasady
Kwas szczawiowy i/lub szczawiany wytrącane są w postaci szczawianu wapnia i rozpuszczane w kwasie siarkowym. Do roztworu dodaje się niewielką ilość roztworu nadmanganianu potasu, który odbarwia się i powoduje tworzenie się dwutlenku węgla. Kiedy powstały dwutlenek węgla przechodzi przez roztwór wodorotlenku baru, tworzy się biały osad (zmętnienie) węglanu baru.
9.2. Procedura
9.2.1. Poddać próbkę przeznaczoną do analizy działaniu opisanemu w punktach od 6.1. do 6.3.; usunie ono każdy obecny środek powierzchniowo czynny.
9.2.2. Dodać na czubku łopatki octanu sodu (4.10) do około 10 ml roztworu otrzymanego zgodnie z p. 9.2.1. i zakwasić roztwór kilkoma kroplami kwasu octowego lodowatego (4.11).
9.2.3. Dodać 10% roztwór chlorku wapnia (4.2) i przesączyć. Rozpuścić osad szczawianu wapnia w 2 ml kwasu siarkowego (1:1) (4.12).
9.2.4. Przenieść roztwór do probówki i dodać kroplami około 0,5 ml 0.1 N roztworu nadmanganianu potasu (4.7) i przesączyć. Jeżeli szczawian jest obecny, roztwór traci kolor najpierw stopniowo, później gwałtownie.
9.2.5. Bezpośrednio po dodaniu nadmanganianu potasu umieścić odpowiednią szklaną rurkę z korkiem nad probówką, ogrzewać lekko zawartość i zbierać powstający dwutlenek węgla w nasyconym roztworze wodorotlenku baru (4.13). Pojawienie się po trzech do pięciu minutach mlecznego zmętnienia węglanu baru świadczy o obecności kwasu szczawiowego.
IV. Oznaczanie chloroformu w paście do zębów
1. Cel i zakres metody
Metoda ta jest stosowana do oznaczania chloroformu w paście do zębów za pomocą chromatografii gazowej. Metoda ta jest odpowiednia do oznaczania chloroformu na poziomie 5% lub poniżej.
2. Definicja
Zawartość chloroformu oznaczona tą metodą jest wyrażona jako procent masowy w wyrobie.
3. Zasady
Otrzymuje się zawiesinę pasty do zębów w mieszaninie dimetyloformamidu/metanolu. do której dodaje się znaną ilość acetonitrylu jako standard wewnętrzny. Po odwirowaniu próby faza ciekła jest analizowana metodą chromatografii gazowej i obliczana jest zawartość chloroformu.
4. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej.
4.1. Porapak Q, Chromosorb 101 lub równoważny, 80 do 100 mesh
4.2. Acetonitryl
4.3. Chloroform
4.4. Dirnetyloformamid
4.5. Metanol
4.6. Roztwór standardu wewnętrznego.
Do 50 ml koibki miarowej dodać pipetą 5 ml dimetyloformamidu (4.4.) i dodać około 300 mg (Mmg) acetonitrylu, dokładnie odważonego. Uzupełnić do kreski dimetyloformamidem i wymieszać.
4.7. Roztwór do oznaczania względnego współczynnika odpowiedzi. Do 10 ml kolbki miarowej dodać pipetą dokładnie 5 ml roztworu wewnętrznego standardu (4.6.) i dodać około 300 mg (M1mg) chloroformu, dokładnie odważonego. Uzupełnić do kreski dimetyloformamidem i wymieszać.
5. Aparatura i wyposażenie
5.1. Waga analityczna
5.2. Chromatograf gazowy z detektorem płomieniowo-jonizacyjnym
5.3. Mikrostrzykawka pojemności 5 do 10 µl i kalibracji co 0,1 µl
5.4. Pipety ze zbiornikiem gruszkowym pojemności 1, 4 i 5 ml
5.5. Kolby miarowe 10 i 50 ml
5.6. Probówki, około 20 ml, z nakrętką, Sovirel France nr 20 lub równoważne. Nakrętka ma wewnętrzną płytkę uszczelniającą, pokrytą z jednej strony teflonem
5.7. Wirówka.
6. Procedura
6.1. Odpowiednie warunki chromatografii gazowej
6.1.1. Materiał kolumny: szkło
| długość: | 150 cm |
| średnica wewnętrzna: | 4 mm |
| średnica zewnętrzna: | 6 mm |
6.1.2. Wypełnić kolumnę wypełnieniem Porapak Q, Chromosorb 101 lub równoważnym 80 do 100 mesh (4.1.) z pomocą wibratora.
6.1.3. Detektor płomieniowo-jonizacyjny: należy nastawić jego czułość tak, aby przy wprowadzeniu 3 µl roztworu według 4.7 wysokość piku acetonitrylu wynosiła około trzech czwartych pełnego zakresu wysokości piku.
6.1.4. Gazy:
Nośnik azot, szybkość przepływu 65 ml/min
Pomocniczy: nastawić przepływ gazów do detektora tak, aby przepływ powietrza lub tlenu był pięć do dziesięciu razy większy niż wodoru.
6.1.5. Temperatury:
| dozownik: | 2100C |
| detektor: | 2100C |
| piec kolumny: | 1750C |
6.1.6. Szybkość przesuwu papieru: około 100 cm na godzinę.
6.2. Przygotowanie próbki
Pobrać próbkę do analizy z nieotwieranej tubki. Usunąć jedną trzecią zawartości, założyć z powrotem nakrętkę na tubę, wymieszać starannie w tubie i następnie pobrać próbkę analityczną.
6.3. Oznaczenie
6.3.1. Do probówki z nakrętką (5.6.) odważyć 6 do 7 g (Mog) z dokładnością do 10 mg pasty do zębów, przygotowanej zgodnie z punktem 6.2, i dodać trzy małe szklane ziarenka.
6.3.2. Do probówki dodać pipetą dokładnie 5 ml roztworu wewnętrznego standardu (4.6.), 4 ml dimetyloformamidu (4.4.) i 1 ml metanolu (4.5.), zamknąć probówkę i wymieszać.
6.3.3. Wytrząsnąć zamkniętą probówkę w mechanicznej wytrząsarce w ciągu pół godziny i odwirować ją w ciągu 15 minut z taką szybkością, aby otrzymać wyraźny rozdział faz. Uwaga: czasami zdarza się, że faza ciekła jest mętna po wirowaniu. Pewną poprawę można uzyskać przez: dodanie 1 do 2 g chlorku sodowego do fazy ciekłej, pozostawienie do odstania i ponowne odwirowanie.
6.3.4. Wprowadzić do chromatografu 3 µl tego roztworu (6.3.3.) w warunkach opisanych w punkcie 6.1. Powtórzyć tę operację w warunkach opisanych powyżej, można podać następujące czasy retencji jako wartości orientacyjne:
| Metanol | średnio 1 minuta |
| Acetonitryl | średnio 2–5 minut |
| Chloroform | średnio 6 minut |
| Dimetyloformamid | >15 minut |
6.3.5. Oznaczenie względnego współczynnika odpowiedzi. Wprowadzić 3 µl roztworu (4.7.) dla oznaczenia tego współczynnika. Powtórzyć operację. Oznaczyć względny współczynnik odpowiedzi codziennie.
7. Obliczenia
7.1. Obliczenie względnej odpowiedzi
7.1.1. Zmierzyć wysokość i szerokość w połowie wysokości pików acetonitrylu i chloroformu i obliczyć powierzchnie obu pików, zgodnie z wzorem: wysokość x szerokość w połowie wysokości.
7.1.2. Oznaczyć powierzchnie pików acetonitrylu i chloroformu w chromatogramie, otrzymanym zgodnie z punktem 6.3.5., i obliczyć względną odpowiedź fs za pomocą następującego wzoru:
| fs = | As x Mi | = | As x 1/10 M |
| Ms x Ai | Ai x M1 |
w którym:
fs – współczynnik względnej odpowiedzi dla chloroformu,
As – powierzchnia piku chloroformu (6.3.5.),
Ai – powierzchnia piku acetonitrylu (6.3.5.),
Ms – ilość chloroformu w mg na 10 ml roztworu omówionego w punkcie 6.3.5. (= M1),
Mi – ilość acetonitrylu w mg na 10 ml roztworu omówionego w punkcie 6.3.5. (= 1/10 M).
Obliczyć średnią z otrzymanych wyników.
7.2. Obliczenie zawartości chloroformu
7.2.1. Obliczyć zgodnie z punktem 7.1.1. powierzchnie pików chloroformu i acetonitrylu na chromatogramie otrzymanym w procedurze opisanej w punkcie 6.3.4.
7.2.2. Obliczyć zawartość chloroformu w paście do zębów za pomocą następującego wzoru:
| %X = | As x Mi | x 100% = | As x M |
| fs x Msx x Ai | fs x Ai x M0 x 100 |
w którym:
%X – zawartość chloroformu w paście do zębów wyrażona jako procent wagowy,
As – powierzchnia piku chloroformu (6.3.5.),
Ai – powierzchnia piku acetonitrylu (6.3.5.),
MSX – masa w mg próbki omówionej w punkcie 6.3.1. (=1 000 x M0),
Mi – ilość acetonitrylu w mg na 10 ml roztworu otrzymanego zgodnie z punktem 6.3.2. (=1/10 M).
Obliczyć średnią oznaczoną zawartość i przedstawić wynik z dokładnością do 0,1%.
8. Powtarzalność
Dla zawartości chloroformu wynoszącej około 3% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć bezwzględnej wartości 0,3%.
V. Oznaczanie cynku
1. Cel i zakres metody
Metoda ta jest odpowiednia do oznaczania cynku w kosmetykach, występującego jako chlorek, siarczan lub 4-hydroksybenzenosulfonian lub jako połączenie kilku z tych soli cynkowych.
2. Definicja
Zawartość cynku w próbce jest oznaczana grawimetrycznie jako bis (tlenek 2-metylo-8-chinolilowy) i wyrażana jako procent masowy cynku w próbce.
3. Zasada
Cynk znajdujący się w roztworze jest w środowisku kwaśnym wytrącany jako bis (tlenek 2-metylo-8-chinolilowo) cynkowy. Po odsączeniu osad suszy się i waży.
4. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej.
4.1. 25% (m/m) stężony amoniak; d420 = 0,91
4.2. Lodowaty kwas octowy
4.3. Octan amonu
4.4. 2-metylochinolin-8-ol
4.5. 6% (m/v) roztwór amoniaku.
Przenieść 240 g stężonego amoniaku (4.1.) do 1 000 ml kolby miarowej, uzupełnić wodą destylowaną do kreski i wymieszać.
4.6. 0,2 M roztwór octanu amonu.
Rozpuścić 15,4 g octanu amonu (4.3.) w wodzie destylowanej w kolbie miarowej 1 000 ml, uzupełnić wodą do kreski i wymieszać.
4.7. Roztwór 2-metylochinolin-8-olu.
Rozpuścić 5 g 2-metylochinolin-8-olu w 12 ml kwasu octowego lodowatego i następnie przenieść z destylowaną wodą do 1 000 ml kolby. Rozcieńczyć wodą do kreski i wymieszać.
5. Aparatura i wyposażenie
5.1. Kolby miarowe, 100 i 1 000 ml
5.2. Zlewki, 400 ml
5.3. Cylindry miarowe, 50 i 150 ml
5.4. Pipety kalibrowane, 10 ml
5.5. Tygle z filtrem G-4
5.6. Kolby próżniowe, 500 ml
5.7. Wodna pompa próżniowa
5.8. Termometr kalibrowany od 0 do 1000C
5.9. Eksykator z odpowiednim środkiem odwadniającym i wskaźnikiem wilgotności, np. żelem krzemionkowym lub równorzędnym
5.10. Piec do suszenia z temperaturą uregulowaną na 150±20C
5.11. Pehametr
5.12. Ogrzewana płytka
5.13. Bibuła filtracyjna, firmy Whatman nr 4 lub równorzędna.
6. Procedura
6.1. Do zlewki pojemności 400 ml zważyć 5 do 10 g (M gramów) analizowanej próbki, zawierającej około 50 do 100 mg cynku, dodać 50 ml wody destylowanej i wymieszać.
6.1.1. Przesączyć, jeśli potrzeba, z pomocą pompy próżniowej, zachować przesącz.
6.1.2. Powtórzyć operację ekstrakcji z dalszymi 50 ml wody destylowanej. Przesączyć i połączyć przesącza.
6.2. Dla każdych 10 mg cynku, znajdujących się w roztworze (6.1.2.), dodać 2 ml roztworu 2-metylochinolin-8-olu (4.7.) i wymieszać.
6.3. Rozcieńczyć mieszaninę 150 ml wody destylowanej, doprowadzić temperaturę mieszaniny do 600C (5.12.) i dodać 45 ml 0,2 M roztworu octanu amonu (4.6.), stale mieszając.
6.4. Doprowadzić pH roztworu do 5,7–5,9, dodawać mieszając 6% roztwór amoniaku (4.5.), pH roztworu zmierzyć pehametrem.
6.5. Odstawić roztwór na 30 minut. Przesączyć z pomocą wodnej pompy próżniowej przez tygiel z filtrem G-4, który wysuszono uprzednio (1500C) i zważono po schłodzeniu (M0 gramów) i przemyć osad 150 ml wody destylowanej o temperaturze 950C.
6.6. Umieścić tygiel z filtrem w piecu suszącym o temperaturze nastawionej na 1500C i suszyć w ciągu jednej godziny.
6.7. Wyjąć filtr z pieca, umieścić w eksykatorze (5.9) i – po schłodzeniu do temperatury pokojowej – oznaczyć masę (M1 gramów).
7. Obliczenie
Obliczyć zawartość cynku w próbce jako procent masowy (% m/m) za pomocą następującego wzoru:
| % cynku = | (M1– M0) x 17,12 |
| M |
w którym:
M – masa w gramach próbki pobranej do analizy według 6.1.,
M0 – masa w gramach pustego i suchego tygla z filtrem (6.5.),
M1 – masa w gramach tygla z filtrem i osadem (6.7.).
8. Powtarzalność
Dla zawartości cynku około 1% (m/m) różnica między wynikami dwu równoległych oznaczeń, wykonywanych dla tej samej próbki, nie powinna przekraczać bezwzględnej wartości 0,1%.
VI. Identyfikacja i oznaczanie kwasu 4-hydroksybenzenosulfonowego
1 Cel i zakres metody
Metoda jest odpowiednia do identyfikacji i oznaczania kwasu 4-hydroksybenzenosulfonowego w kosmetykach takich, jak aerozole i płyny do twarzy.
2. Definicja
Zawartość kwasu 4-hydroksybenzenosulfonowego, oznaczona zgodnie z tą metodą, jest wyrażona jako procent wagowy bezwodnego 4-hydroksybenzenosulfonianu cynku w wyrobie.
3. Zasada
Próbka analityczna zostaje zatężona pod zmniejszonym ciśnieniem, rozpuszczona w wodzie i oczyszczona przez ekstrakcję chloroformem. Kwas 4-hydroksybenzenosulfonowy oznacza się jodometrycznie na części przesączonego roztworu wodnego.
4. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej.
4.1. 36% (m/m) stężony kwas chlorowodorowy d420 = 1,18
4.2. Chloroform
4.3. Butan-1-ol
4.4. Kwas octowy lodowaty
4.5. Jodek potasu
4.6. Bromek potasu
4.7. Węglan sodu
4.8. Kwas sulfanililowy
4.9. Azotyn sodu
4.10. 0,1 N bromian potasu
4.11. 0,1 N roztwór tiosiarczanu sodu
4.12. 1% (m/v) wodny roztwór skrobi
4.13. 2% (m/v) wodny roztwór węglanu sodu
4.14. 4,5% (m/v) wodny roztwór azotynu sodu
4.15. 0,05% (m/v) roztwór ditizonu w chloroformie
4.16. Roztwór rozwijający: butan-1-ol/kwas octowy lodowaty/woda (4:1:5 części objętościowych); po zmieszaniu w rozdzielaczu oddzielić dolną fazę
4.17. Odczynnik Pauly'ego: rozpuścić 4,5 g kwasu sulfanililowego (4.8.) w 45 ml stężonego kwasu chlorowodorowego (4.1.) ogrzewając i rozcieńczyć roztwór wodą do 500 ml. Schłodzić 10 ml roztworu w naczyniu z wodą i lodem i dodać, mieszając, 10 ml zimnego roztworu azotynu sodu (4.14.). Odstawić roztwór na 15 minut w temperaturze 00C (w tej temperaturze roztwór jest trwały w ciągu jednego do trzech dni) i – bezpośrednio przed spryskiwaniem (7.5.) – dodać 20 ml roztworu węglanu sodu (4.13.).
4.18. Gotowe przygotowane płytki celulozowe do chromatografii cienkowarstwowej: wielkość 20 x 20 cm, grubość warstwy adsorbenta 0,25 mm.
5. Aparatura i wyposażenie
5.1. Okrągłodenne kolby ze szlifowanym korkiem szklanym, 100 ml
5.2. Rozdzielacz, 100 ml
5.3. Kolba stożkowa ze szlifowanym korkiem szklanym, 250 ml
5.4. Biureta, 25 ml
5.5. Pipety ze zbiornikiem gruszkowym, 1, 2 i 10 ml
5.6. Pipeta kalibrowana, 5 ml
5.7. Mikrostrzykawka, 10 µl z kalibracją co 0,1 µl
5.8. Termometr kalibrowany od 0 do 1000C
5.9. Łaźnia wodna z elementem grzewczym
5.10. Piec do suszenia, dobrze przewietrzany i nastawiony na temperaturę 800C
5.11. Typowe urządzenie do wykonania chromatografii cienkowarstwowej.
6. Przygotowanie próbki
W opisanej poniżej metodzie identyfikacji i oznaczania kwasu hydroksybenzenosulfonowego w aerozolach używana jest pozostałość otrzymana przez usunięcie z pojemnika aerozolowego rozpuszczalników i propelentów, które wyparowują pod normalnym ciśnieniem.
7. Identyfikacja
7.1. Nanieść za pomocą mikrostrzykawki (5.7.) po 5 µl pozostałości (6) lub próbki w każdym z sześciu punktów na linii początkowej w odległości 1 cm od dolnej krawędzi płytki do chromatografii cienkowarstwowej (4.18.).
7.2. Umieścić płytkę w komorze rozwijającej, która już zawiera roztwór rozwijający (4.16.) i rozwijać do osiągnięcia przez czoło rozpuszczalnika odległości 15 cm od linii początkowej.
7.3. Wyjąć płytkę z roztworu rozwijającego i wysuszyć w 800C do momentu, kiedy opary kwasu octowego nie będą wyczuwalne. Spryskać płytkę roztworem węglanu sodu (4.13.) i wysuszyć na powietrzu.
7.4. Przykryć połowę płytki szklaną płytką i spryskać nieprzykrytą część 0,05% roztworem ditizonu (4.15.). Pojawienie się purpurowo-czerwonych plam na chromatogramie wskazuje na obecność jonów cynku.
7.5. Przykryć napryskaną część płytki szklaną płytką i spryskać pozostałą część odczynnikiem Pauly'ego (4.17.). Na obecność kwasu 4-hydroksybenzenosulfonowego wskazuje pojawienie się żółto-brązowych plam o wartości Rf około 0,26, podczas gdy żółta plama o wartości Rf około 0,45 na chromatogramie wskazuje na obecność kwasu 3-hydroksybenzenosulfonowego.
8. Oznaczenie
8.1. Do okrągłodennej kolby pojemności 100 ml odważyć 10 g próbki lub pozostałości (6) i odparować prawie do sucha pod próżnią w wyparce obrotowej na łaźni wodnej o temperaturze 400C.
8.2. Wprowadzić pipetą do kolby 10 ml (V1 ml) wody i rozpuścić pozostałość po odparowaniu (8.1.) przez ogrzewanie.
8.3. Ilościowo przenieść roztwór do rozdzielacza (5.2.) i ekstrahować wodny roztwór dwukrotnie porcjami po 20 ml chloroformu (4.2.). Po każdej ekstrakcji odrzucać fazę chloroformową.
8.4. Przesączyć wodny roztwór przez karbowany sączek. Zależnie od oczekiwanej zawartości kwasu hydroksybenzenosulfonowego przenieść pipetą 1,0 lub 2 ml (V2) przesączu do 250 ml kolby stożkowej (5.3.) i rozcieńczyć wodą do 75 ml.
8.5. Dodać 2,5 ml 36% kwasu chlorowodorowego (4.1.) i 2,5 g bromku potasu (4.6.), zmieszać i doprowadzić temperaturę roztworu do 500C z pomocą łaźni wodnej.
8.6. Dodawać z biurety 0,1 N roztworu bromianu potasu (4.10.) aż roztwór, stale o temperaturze 500C, stanie się żółty.
8.7. Dodać dalsze 3,0 ml roztworu bromianu potasu (4.10.), zamknąć kolbę korkiem i pozostawić na 10 minut w łaźni wodnej o temperaturze 500C. Jeśli po 10 minutach roztwór straci swoją barwę, dodać dalsze 2,0 ml roztworu bromianu potasu (4.10.), zamknąć kolbę korkiem i ogrzewać przez 10 minut w łaźni wodnej, utrzymywanej w temperaturze 500C. Zanotować całkowitą ilość dodanego roztworu bromianu potasu (a).
8.8. Schłodzić roztwór do temperatury pokojowej, dodać 2 g jodku potasu (4.5.) i wymieszać.
8.9. Odmiareczkować powstały jod 0,1 N roztworem tiosiarczanu sodu (4.11.). Przy końcu miareczkowania dodać kilka kropli roztworu skrobi (4.12.) jako wskaźnika. Zanotować ilość użytego tiosiarczanu sodu (b).
9. Obliczenie
Obliczyć zawartość hydroksybenzenosulfonianu cynku w próbce lub pozostałości (6) jako procent masowy (m/m) za pomocą następującego wzoru:
| % (m/m) hydroksybenzenosulfonianu cynku = | (a – b) x V1 x 0,00514 x 100 | |
| m x V2 |
| |
w którym:
a – całkowita ilość w mililitrach dodanego 0,1 N roztworu bromianu potasu (8.7.),
b – ilość w mililitrach 0,1 N roztworu tiosiarczanu sodu (4.11.) użytego do odmiareczkowania (8.9.),
m – ilość analizowanego produktu lub pozostałości wyrażona w miligramach (8.1.),
V1 – objętość roztworu otrzymanego według 8.2. wyrażona w mililitrach,
V2 – objętość rozpuszczonej pozostałości po odparowaniu użytej do analizy (8.4.) wyrażona w mililitrach.
W przypadku aerozoli wynik pomiaru w % (m/m) pozostałości (6) musi być wyrażony w odniesieniu do oryginalnego wyrobu. W celu wykonania tej zamiany podano zasady pobierania próbek aerozoli.
10. Powtarzalność
Dla zawartości około 5% hydroksybenzenosulfonianu cynku różnica między wynikami dwu oznaczeń, przeprowadzonych równolegle dla tej samej próbki, nie powinna przekraczać bezwzględnej wartości 0,5%.
11. Interpretacja wyników
Maksymalna zawartość 4-hydroksybenzenosulfonianu cynku w płynach do twarzy i dezodorantach wynosi 6% (m/m). Sformułowanie to oznacza, że obok zawartości kwasu hydroksybenzenosulfonowego należy oznaczyć zawartość cynku. Pomnożenie obliczonej zawartości hydroksybenzenosulfonianu cynku (9) przez współczynnik 0,1588 daje minimalną zawartość cynku w % (m/m), która teoretycznie musi się znajdować w wyrobie w związku z oznaczoną zawartością kwasu hydroksybenzenosulfonowego. Zawartość cynku – rzeczywiście oznaczona grawimetrycznie – może jednak być wyższa, ponieważ chlorek cynku i siarczan cynku mogą być również używane w kosmetykach.
VII. Identyfikowanie środków utleniających i oznaczanie nadtlenku wodoru w kosmetykach do pielęgnacji włosów
CEL l ZAKRES
Jodometryczne oznaczanie nadtlenku wodoru w kosmetykach jest możliwe jedynie w przypadku nieobecności innych środków utleniających, które uwalniają jod z jodków. W konsekwencji konieczne jest przed jodometrycznym oznaczaniem nadtlenku wodoru wykrycie i zidentyfikowanie innych obecnych środków utleniających. Identyfikacja ta dzieli się na dwa etapy: pierwszy obejmuje nadsiarczany, bromiany i nadtlenek wodoru, a drugi nadtlenek baru.
A. IDENTYFIKOWANIE NADSIARCZANÓW, BROMIANÓW l NADTLENKU WODORU
1. Zasada
Nadsiarczan sodu, nadsiarczan potasu i nadsiarczan amonu oraz bromian potasu, bromian sodu i nadtlenek wodoru – pochodzące lub nie z nadtlenku baru – są identyfikowane przez bibułową chromatografię zstępującą, z zastosowaniem dwóch rozpuszczalników rozwijających.
2. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej.
2.1 0,5% (m/v) wodne roztwory następujących związków:
2.1.1. Nadsiarczan sodu
2.1.2. Nadsiarczan potasu
2.1.3. Nadsiarczan amonu
2.1.4. Bromian potasu
2.1.5. Bromian sodu
2.1.6. Nadtlenek wodoru
2.2. Rozpuszczalnik rozwijający A, etanol 80% (v/v)
2.3. Rozpuszczalnik rozwijający B, benzen - metanol -3-metylobutan-1-ol -woda (34:38:18:10 obj.)
2.4. Środek wywołujący A, 10% (m/v) wodny roztwór jodku potasu
2.5. Środek wywołujący B, 1% (m/v) wodny roztwór skrobi
2.6. Środek wywołujący C, 10% (m/m) kwas solny
2.7. 4 N kwas solny
3. Aparatura i wyposażenie
3.1. Bibuła chromatograficzna (Whatman Nr 3 i 4 lub ich równoważniki)
3.2. Mikropipeta, 1 µl
3.3. Kolba miarowa, 100 ml
3.4. Sączki karbowane
3.5. Aparatura do zstępującej chromatografii bibułowej
4. Przygotowanie próbki
4.1. Wyroby rozpuszczalne w wodzie
Przygotować po dwa roztwory każdej próbki przez rozpuszczenie 1 g i 5 g wyrobu kolejno w 100 ml wody. Pobrać po 1 ml każdego z tych roztworów do analizy metodą chromatografii bibułowej opisaną w punkcie 5.
4.2. Wyroby słabo rozpuszczalne w wodzie
Odważyć 1 g i 5 g próbki i zawiesić w 50 ml wody, uzupełnić do 100 ml wodą w obu przypadkach i wymieszać. Przesączyć obie dyspersje przez karbowany lejek (3.4.) i pobrać 1 µl każdego filtratu do analizy metodą chromatografii bibułowej opisanej w punkcie 5.
4.3. Kremy
Zawiesić 5 g i 20 g każdego wyrobu w 100 ml wody i zastosować dyspersje do wykonania analizy metodą chromatografii bibułowej opisaną w punkcie 5.
5. Metoda
5.1. Umieścić odpowiednią ilość rozpuszczalników A (2.2.) i B (2.3.) w dwóch oddzielnych komorach chromatograficznych w celu przeprowadzenia zstępującej chromatografii bibułowej. Nasycać komory chromatograficzne oparami rozpuszczalników w ciągu co najmniej 24 godzin.
5.2. Nanieść po 1 µl jednego roztworu próbki i jednego roztworu odniesienia przygotowanych według opisu w punkcie 4 i 2.1. w każdym punkcie startowym paska bibuły chromatograficznej (Whatman Nr 3 lub równoważnik) długości 40 cm i szerokości 20 cm (3.1.) lub innego odpowiedniego formatu i odparować rozpuszczalnik na powietrzu.
5.3. Umieścić pasek chromatograficzny (5.2.) w komorze chromatograficznej z rozpuszczalnikiem rozwijającym A (5.1.) i rozwijać do chwili, kiedy czoło rozpuszczalnika osiągnie odległość 35 cm (około 15 godzin).
5.4. Powtórzyć procedurę opisaną w punkcie 5.2. i 5.3. z użyciem bibuły chromatograficznej (Whatman Nr 4 lub równoważnik) (3.1.) i rozpuszczalnikiem rozwijającym B. Rozdział chromatograficzny prowadzić do chwili, kiedy czoło rozpuszczalnika osiągnie odległość 35 cm (około pięciu godzin).
5.5. Po rozwinięciu wyjąć chromatogramy z komory i wysuszyć na powietrzu.
5.6. Wywołać plamy na chromatogramie przez spryskanie go kolejno:
5.6.1. Środkiem wywołującym A (2.4.) i następnie po krótkim czasie środkiem wywołującym B (2.5.). Plamy nadsiarczanów pojawią się pierwsze na chromatogramie, a następnie wywołane zostają plamy nadtlenku wodoru.
Obrysować plamy ołówkiem.
5.6.2. Środkiem wywołującym C (2.6.) chromatogramy otrzymane zgodnie z opisem w punkcie 5.6.1.; szaroniebieskie plamy na chromatogramie wskażą na obecność bromianów.
5.7. W opisanych powyżej warunkach właściwych dla rozpuszczalników rozwijających A (2.2.) i B (2.3.), wartości Rf dla substancji odniesienia (2.1.) są w przybliżeniu następujące:
| roztwór rozwijający A | (2.2.) | roztwór rozwijający B (2.3.) | |
| nadsiarczan sodu | 0,40 |
| 0,10 |
| nadsiarczan potasu | 0,40 |
| 0,02 + 0,05 |
| nadsiarczan amonu | 0,50 |
| 0,10 + 0,20 |
| bromian sodu | 0,40 |
| 0,20 |
| bromian potasu | 0,40 |
| 0,10 + 0,20 |
| nadtlenek wodoru | 0,80 |
| 0,80 |
B. IDENTYFIKOWANIE NADTLENKU BARU
1. Zasada
Nadtlenek baru jest identyfikowany na podstawie powstawania nadtlenku wodoru po zakwaszeniu próbki (A.4.2.) i obecności jonu baru:
– w nieobecności nadsiarczanów (A) przez dodanie rozcieńczonego kwasu siarkowego do części kwaśnego roztworu próbki (B.4.1.), w wyniku czego powstaje biały osad siarczanu baru. Obecność jonów baru w próbce (B.4.1.) jest ponownie potwierdzona metodą chromatografii bibułowej według metody opisanej poniżej (B.5.),
– jeśli równocześnie w próbce obecne są nadtlenek baru i nadsiarczany (B.4.2.) przez poddanie stapianiu pozostałości z roztworu (B.4.2.) w środowisku alkalicznym, po rozpuszczeniu w kwasie solnym, obecność jonów baru w roztworze stopu (B.4.2.3.) stwierdza się metodą chromatografii bibułowej i/lub przez wytrącenie osadu siarczanu baru.
2. Odczynniki
2.1. Metanol
2.2. 36% (m/m) stężony kwas solny
2.3. 6 N kwas solny
2.4. 4 N kwas siarkowy
2.5. Sól sodowa kwasu rodyzonowego (3,4,5,6-tetraoksocykloheksen-1,2-diolu)
2.6. Chlorek baru (BaCI2 x 2H2O)
2.7. Bezwodny węglan sodu
2.8. 1% (m/v) wodny roztwór chlorku baru
2.9. Rozpuszczalnik rozwijający zawierający metanol, stężony kwas solny (stężenie 36%) i wodę (80:10:10 obj.)
2.10. Środek wywołujący, 0,1% (m/v) wodny roztwór soli disodowej kwasu rodyzonowego, świeżo przygotowany bezpośrednio przed użyciem.
3. Aparatura i wyposażenie
3.1. Mikropipeta, 5 µl
3.2. Tygle platynowe
3.3. Kolby miarowe, 100 ml
3.4. Bibuła chromatograficzna firmy Schleicher and Schul I 2043b lub równoważna. Bibułę oczyszcza się przez pozostawienie jej na noc w komorze chromatograficznej (A.3.5.) zawierającej rozpuszczalnik rozwijający (B.2.9.) i następnie suszy.
3.5. Karbowany sączek bibułowy
3.6. Aparatura do zstępującej chromatografii bibułowej
4. Przygotowanie próbki
4.1. Wyroby niezawierające nadsiarczanów
4.1.1. Zawiesić 2 g wyrobu w 50 ml wody i doprowadzić kwasem solnym (B.2.3.) pH dyspersji do wartości około 1.
4.1.2. Za pomocą wody przenieść dyspersję do 100 ml kolby miarowej, uzupełnić wodą do kreski i wymieszać. Używać tej dyspersji do analizy metodą chromatografii bibułowej opisanej w punkcie 5 i do identyfikowania obecności baru przez wytrącanie siarczanu.
4.2. Wyroby zawierające nadsiarczany.
4.2.1. Zawiesić 2 g wyrobu w 100 ml wody i przesączyć.
4.2.2. Do wysuszonej pozostałości dodać siedem do dziesięciu razy więcej wagowo węglanu sodu (B.2.7.), wymieszać i stapiać mieszaninę w tyglu platynowym (B.3.2.) w ciągu pół godziny.
4.2.3. Schłodzić do temperatury pokojowej, rozpuścić stop w 50 ml wody i przesączyć (B.3.5.).
4.2.4. Rozpuścić pozostałość ze stopu w kwasie solnym (B.2.3.) i uzupełnić wodą do 100 ml. Używać tego roztworu do analizy metodą chromatografii bibułowej opisaną w punkcie 5 i do identyfikowania obecności baru przez wytrącanie siarczanu.
5. Metoda
5.1. Umieścić odpowiednią ilość rozpuszczalnika rozwijającego (B.2.9.) w komorze do zstępującej chromatografii bibułowej i wysycać komorę w ciągu co najmniej 15 godzin.
5.2. Na kawałku bibuły chromatograficznej – przygotowanej tak jak opisano w punkcie B.3.4. – nanosić po 5 µl każdego roztworu przygotowanego według punktu B.4.1.2. i B.4.2.4. i roztworu odniesienia B.2.8. w trzech punktach startowych.
5.3. Wysuszyć bibułę z naniesionymi próbkami i roztworem odniesienia na powietrzu. Rozwijać chromatogram do chwili, kiedy czoło rozpuszczalnika osiągnie 30 cm.
5.4. Wyjąć chromatogram z komory i wysuszyć na powietrzu.
5.5. Wywołać plamy na chromatogramie przez spryskanie bibuły środkiem wywołującym (B.2.10,). W przypadku obecności baru pojawiają się na chromatogramie czerwone plamy o wartości Rf około 0,10.
C. OZNACZANIE NADTLENKU WODORU
1. Zasada
Jodometryczne oznaczanie nadtlenku wodoru polega na następującej reakcji:
H2O2 + 2H+ + 2Jˉ → J2 + 2H2O
Przemiana ta zachodzi powoli, lecz można ją przyspieszyć przez dodatek molibdenianu amonu. Ilość powstałego jodu jest oznaczona miareczkowo tiosiarczanem sodu i stanowi miarę zawartości nadtlenku wodoru.
2. Definicja
Zawartość nadtlenku wodoru oznaczona w sposób opisany poniżej jest wyrażona jako procent masowy (% m/m) wyrobu.
3. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej
3.1. 2 N kwas siarkowy
3.2. Jodek potasu
3.3. Molibdenian amonu
3.4. 0,1 N tiosiarczan sodu
3.5. 10% (m/v) roztwór jodku potasu, świeżo przygotowany bezpośrednio przed użyciem
3.6. 20% (m/v) roztwór molibdenianu amonu
3.7. 1% (m/v) roztwór skrobi
4. Aparatura i wyposażenie
4.1. Zlewki, 100 ml
4.2. Biureta, 50 ml
4.3. Kolby miarowe, 250 ml
4.4. Cylindry miarowe, 25 i 100 ml
4.5. Pipety z jednym oznaczeniem objętości, 10 ml
4.6. Kolby stożkowe, 250 ml
5. Metoda
5.1. Odważyć około 10 g (m gramów) wyrobu zawierającego około 0,6 g nadtlenku wodoru w zlewce 100 ml. Przenieść zawartość zlewki z pomocą wody do kolby miarowej 250 ml, uzupełnić wodą do kreski i wymieszać.
5.2. Odmierzyć pipetą 10 ml roztworu próbki (5.1.) do 250 ml kolby stożkowej (4.6.) i dodawać kolejno 100 ml 2 N kwasu siarkowego (3.1.), 20 ml roztworu jodku potasu (3.5.) i trzy krople roztworu molibdenianu amonu (3.6.).
5.3. Odmiareczkować powstały jod bezzwłocznie 0,1 N roztworem tiosiarczanu sodu (3.4.) i bezpośrednio przed osiągnięciem punktu końcowego dodać kilka mililitrów roztworu skrobi jako wskaźnika (3.7.). Zarejestrować zużycie 0,1 N roztworu tiosiarczanu sodu (3.4.) w mililitrach (V).
5.4. Wykonać według sposobu opisanego w punkcie 5.2. i 5.3. analizę ślepej próby, zastępując 10 ml roztworu próbki 10 ml wody. Zarejestrować zużycie 0,1 N roztworu tiosiarczanu sodu w analizie ślepej próby (V0 ml).
6. Obliczenie
Obliczyć zawartość nadtlenku wodoru w wyrobie w procentach masowych (% m/m) według następującego wzoru:
| % nadtlenku wodoru = | (V – V0) x 1,7008 x 250 x 100 | = | (V–V0) x 4,252 |
| m x 10 x 1 000 | m |
w którym:
m – ilość analizowanego wyrobu (5.1.),
V0 – zużycie 0,1 N roztworu tiosiarczanu sodu do analizy ślepej próby w mililitrach (5.4.),
V – zużycie 0,1 N roztworu tiosiarczanu sodu do analizy roztworu próbki w mililitrach (5.3.).
7. Powtarzalność
Dla wyrobu zawierającego około 6% (m/m) nadtlenku wodoru różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych równolegle dla tej samej próbki nie powinna przekraczać wartości bezwzględnej 0,2%.
VIII. Identyfikowanie i oznaczanie azotynów
A. IDENTYFIKOWANIE
1. Cel i zakres
Metoda jest odpowiednia do identyfikowania azotynu w kosmetykach, szczególnie w kremach i pastach.
2. Zasada
Na obecność azotynu wskazuje powstawanie barwnych pochodnych z fenylohydrazonem aldehydu 2-aminobenzoesowego (Nitrin®).
3. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej.
3.1. Rozcieńczony kwas siarkowy; rozcieńczyć 2 ml stężonego kwasu siarkowego (d420 = 1,84) w 11 ml wody destylowanej.
3.2. Rozcieńczony kwas solny: rozcieńczyć 1 ml stężonego kwasu solnego (d420 = 1,19) w 11 ml wody destylowanej.
3.3. Metanol
3.4. Roztwór fenylohydrazonu aldehydu 2-aminobenzoesowego (odczynnik Nitrin(®) w metanolu. Odważyć 2,0 g Nitrinu® i przenieść ilościowo do 100 ml kolby miarowej. Dodać kroplami 4 ml rozcieńczonego kwasu solnego (3.2.) i wytrząsać. Uzupełnić do kreski metanolem i mieszać do chwili, aż roztwór stanie się całkowicie przezroczysty. Przechowywać roztwór w brązowej szklanej butli (4.3.).
4. Aparatura
4.1. Zlewki, 50 ml
4.2. Kolba miarowa, 100 ml
4.3. Butla z brązowego szkła, 125 ml
4.4. Płytka szklana, 10 x 10 cm
4.5. Łopatka z tworzywa sztucznego
4.6. Bibuła filtracyjna, 10 x 10 cm
5. Procedura
5.1. Rozsmarować część badanej próbki równomiernie na płytce szklanej (4.4.), tak aby pokryć powierzchnię do grubości nie większej niż 1 cm.
5.2. Zanurzyć część arkusza bibuły filtracyjnej (4.6.) w wodzie destylowanej. Położyć ją na próbce i przycisnąć bibułę filtracyjną łopatką z tworzywa sztucznego (4.5.).
5.3. Odczekać około jednej minuty i nanieść na środek bibuły filtracyjnej:
Dwie krople rozcieńczonego kwasu siarkowego (3.1.) i następnie dwie krople roztworu Nitrinu® (3.4.).
5.4. Po 5 do 10 sekundach zdjąć bibułę filtracyjną i obejrzeć ją w świetle dziennym. Czerwonawopurpurowe zabarwienie wskazuje na obecność azotynu. Jeśli zawartość azotynu jest niewielka, czerwonawopurpurowa barwa zmienia się w żółtą po 5 do 15 sekundach. Jeśli obecne są duże ilości azotynu, taka zmiana barwy zachodzi dopiero po 1 do 2 minut.
6. Intensywność czerwonawopurpurowej barwy i czas, który upływa przed jej zmianą na żółtą może być wskazówką do oceny zawartości azotynu w próbce.
B. OZNACZANIE AZOTYNÓW
1. Cel
Metoda opisuje znaczenie azotynów w kosmetykach
2. Definicja
Zawartość azotynów w próbce, oznaczona zgodnie z tą metodą, jest wyrażona w % masowych azotynu sodu.
3. Zasada
Po rozcieńczeniu próbki wodą i wyklarowaniu roztworu azotyn obecny w próbce jest poddany reakcji z sulfaniloamidem i N-1-naftyloetylenodiaminą i oznacza się gęstość optyczną otrzymanej barwy przy 538 nm.
4. Odczynniki
Wszystkie odczynniki powinny być czystości analitycznej.
4.1. Odczynniki klarujące odczynniki te nie mogą być używane dłużej niż w ciągu tygodnia po przygotowaniu:
4.1.1. Odczynnik l Carreza:
Rozpuścić 106 g cyjanożelazianu potasu (II), K4Fe(CN)6 x 3 H2O w wodzie destylowanej i rozcieńczyć wodą do 1 000 ml.
4.1.2. Odczynnik II Carreza:
Rozpuścić 219,5 g octanu cynku, Zn(Ch3COO)2 x 2 H2O i 30 ml lodowatego kwasu octowego w wodzie destylowanej i rozcieńczyć wodą do 1 000 ml.
4.2. Roztwór azotynu sodu:
Rozpuścić 0,500 g azotynu sodu w wodzie destylowanej w kolbie miarowej 1 000 ml i rozcieńczyć wodą do kreski, rozcieńczyć 10,0 ml tego bazowego standardowego roztworu do 500 ml; 1 ml tego roztworu = 10 mikrogramów NaNO2.
4.3. 1N roztwór wodorotlenku sodu
4.4. 0,2% chlorowodorku sulfaniloamidu: rozpuścić 2,0 g sulfaniloamidu w 800 ml wody podczas ogrzewania. Schłodzić i dodać mieszając 100 ml stężonego kwasu solnego. Rozcieńczyć wodą do 1 000 ml.
4.5. 5 N kwas solny
4.6. Odczynnik N-1-naftylowy
Ten roztwór musi być przygotowany w dniu zastosowania. Rozpuścić 0,1 g chlorowodorku N-1-naftylenodiaminy w wodzie i rozcieńczyć wodą do 100 ml.
5. Aparatura
5.1. Waga analityczna
5.2. Kolby miarowe 100, 250, 500 i 1 000 ml
5.3. Pipety ze zbiornikiem lub kalibrowane
5.4. Cylindry miarowe 100 ml
5.5. Sączki filtracyjne karbowane o średnicy 15 cm, niezawierające azotynów
5.6. Łaźnia wodna
5.7. Spektrofotometr z naczyńkami pomiarowymi o długości drogi optycznej 1 cm
5.8. Pehametr
5.9. Mikrobiureta, 10 ml
5.10. Zlewki, 250 ml.
6. Procedura
6.1. Odważyć około 0,5 g (m gramów) z dokładnością do 0,1 mg homogenizowanej próbki, przenieść z pomocą gorącej wody destylowanej ilościowo do 250 ml zlewki (5.10.) i uzupełnić objętość gorącą wodą destylowaną do około 150 ml. Umieścić zlewkę (5.10) w łaźni wodnej (5.6.) o temperaturze 800C na pół godziny. Podczas tego okresu od czasu do czasu potrząsać zlewkę.
6.2. Schłodzić do temperatury pokojowej i dodawać kolejno, podczas mieszania, 2 ml odczynnika l Carreza (4.1.1.) i 2 ml odczynnika II Carreza (4.1.2.).
6.3. Dodawać 1 N roztwór wodorotlenku sodu (4.3.) aż do uzyskania pH 8,3 (Używać pehametru (5.8.)). Przenieść zawartość ilościowo do 250 ml kolby miarowej (5.2.) i uzupełnić do kreski wodą destylowaną.
6.4. Wymieszać zawartość i przesączyć przez sączek karbowany (5.5).
6.5. Przenieść pipetą (5.3.) odpowiednią ilość (V ml) klarownego przesączu, lecz nie więcej niż 25 ml, do 100 ml kolby miarowej (5.2.) i dodać wody destylowanej do objętości 60 ml.
6.6. Po zmieszaniu dodać 10,0 ml roztworu chlorowodorku sulfaniloamidu (4.4.) i następnie 6,0 ml 5N roztworu kwasu solnego (4.5.). Wymieszać i pozostawić do odstania na pięć minut. Dodać 2 ml odczynnika N-1-naftylowego (4.6.), wymieszać i pozostawić do odstania na trzy minuty. Rozcieńczyć wodą do kreski i wymieszać.
6.7. Przygotować ślepą próbę, powtarzając operację 6.5. i 6.6. bez dodawania odczynnika N-1-naftylowego (4.6.).
6.8. Zmierzyć za pomocą spektrofotometru (5.7.) gęstość optyczną przy 538 nm roztworu otrzymanego według punktu 6.6, używając ślepej próby (6.7.) jako odnośnika.
6.9. Odczytać z wykresu kalibracyjnego (6.10.) zawartość azotynu sodu w mikrogramach na 100 ml roztworu (m1) mikrogramów), która odpowiada gęstości optycznej zmierzonej w punkcie 6.8.
6.10. Przygotować wykres kalibracyjny dla stężeń 0,20, 40, 60, 80, 100 µg azotynu sodu w 100 ml, stosując roztwór azotynu sodowego o stężeniu 10 µg na ml (4.2.).
7. Obliczenia
Obliczyć zawartość azotynu sodu w procentach masowych za pomocą następującego wzoru:
| %(m/m) NaNO2 | = | 250 | x m1 x 10–6 x | 100 | = | m1 |
| V | m | V x m x 40 |
w którym:
m – masa póbki pobranej do analizy w gramach,
m1 – zawartość azotynu sodu w mikrogramach oznaczona w punkcie 6.9,
V – ilość m1 przesączu użytego do pomiaru (6.5.).
8. Powtarzalność
Dla zawartości około 0,2% m/m azotynu sodu różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć wartości absolutnej 0,005%.
IX. Identyfikacja i oznaczanie wolnych wodorotlenków sodu i potasu
1. Cel i zakres metody
Metoda podaje dokładną procedurę identyfikacji kosmetyków zawierających znaczne ilości wodorotlenków sodu i/lub potasu i oznaczania takich wolnych wodorotlenków sodu i/lub potasu w środkach do prostowania włosów i rozpuszczalnikowych środkach do usuwania skórek paznokci.
2. Definicja
Wolny wodorotlenek sodu i potasu są określane objętością (ilością) standardowego roztworu kwasu, wymaganą do zobojętnienia produktu w podanych warunkach; otrzymana ilość wyraża się jako % m/m wolnego wodorotlenku.
3. Zasady
Próbka zostaje rozpuszczona lub zdyspergowana w wodzie i miareczkowana standardowym roztworem kwasu. Wartość pH jest rejestrowana równocześnie z dodawaniem kwasu do roztworu wodorotlenku sodu lub potasu: punktem końcowym jest wyraźny wzrost szybkości zmian wartości pH. Krzywa miareczkowania może być niewyraźna w obecności:
(a) amoniaku lub innych słabych zasad organicznych, które same mają raczej spłaszczoną krzywą miareczkowania z kilkoma punktami przegięcia. W tej metodzie amoniak usuwa się przez odparowanie pod zmniejszonym ciśnieniem, ale w pokojowej temperaturze,
(b) soli słabych kwasów, które mogą powodować wzrost krzywej miareczkowania z kilkoma punktami przegięcia. W tych przypadkach tylko pierwsza część krzywej do pierwszego z tych punktów przegięcia odpowiada zobojętnianiu jonu wodorotlenowego pochodzącego z wolnego wodorotlenku sodu lub potasu. Podano inną procedurę miareczkowania, odpowiednią dla przypadku, gdy zauważa się nadmierne oddziaływanie soli słabych kwasów nieorganicznych. Chociaż istnieje teoretyczna możliwość, że inne rozpuszczalne mocne zasady, np. wodorotlenek litu, czwartorzędowy wodorotlenek amonu, mogłyby być obecne, dając wzrost pH do wysokich wartości, jednak obecność ich w tym rodzaju kosmetyków jest wysoce nieprawdopodobna.
4. Identyfikowanie
4.1. Odczynniki
4.1.1. Standardowy alkaliczny roztwór buforowy o pH 9.18 w 250C 0.05M roztwór tetraboranu sodu.
4.2. Aparatura
4.2.1. Zwykłe szklane wyposażenie laboratoryjne
4.2.2. Pehametr
4.2.3. Szklana elektroda membranowa
4.2.4. Standardowa kalomelowa elektroda odniesienia
4.3. Procedura
Skalibrować pehametr za pomocą elektrod z zastosowaniem standardowego roztworu buforowego. Przygotować 10% roztwór lub dyspersję analizowanego wyrobu w wodzie i przesączyć go. Zmierzyć pH. Jeżeli pH wynosi 12 lub więcej, należy wykonać ilościowe oznaczenie.
5. Oznaczanie
5.1. Miareczkowanie w środowisku wodnym
5.1.1. Odczynnik
5.1.1.1. Standardowy 0.1 N kwas chlorowodorowy
5.1.2. Aparatura
5.1.2.1. Zwykłe szklane wyposażenie laboratoryjne
5.1.2.2. Pehametr, korzystnie, jeśli wyposażony w rejestrator
5.1.2.3. Szklana elektroda membranowa
5.1.2.4. Standardowa kalomelowa elektroda odniesienia
5.1.3. Procedura
Zwarzyć dokładnie w zlewce o pojemności 150 ml próbkę o wielkości 0.5–1.0 g. Jeżeli w próbce znajduje się amoniak, dodać kilka ziaren porowatych, umieścić zlewkę w eksykatorze próżniowym, usuwać amoniak za pomocą pompy wodnej tak długo, aż zapach jego będzie niewyczuwalny (około trzech godzin). Dodać 100 ml wody, rozpuścić lub zdyspergować pozostałość i miareczkować 0.1 N roztworem kwasu chlorowodorowego (5.1.1.1.), rejestrując zmiany pH (5.1.2.2.).
5.1.4. Obliczanie
Umiejscowić punkty przegięcia na krzywych miareczkowania. Jeżeli pierwszy punkt przegięcia występuje przy pH niższym niż 7, próbka nie zawiera wodorotlenku sodu lub potasu. Jeżeli na krzywej znajdują się dwa lub więcej punktów przegięcia, tylko pierwszy punkt ma znaczenie. Zanotować objętość roztworu zużytego do miareczkowania do pierwszego punktu przegięcia.
Oznaczyć symbolem V objętość roztworu zużytego do miareczkowania, w ml.
Oznaczyć symbolem M masę próbki analitycznej, w gramach. Zawartość wodorotlenku sodu i/lub potasu w próbce wyrażoną jako % (m/m) wodorotlenku sodu oblicza się, stosując wzór:
| % (m/m) = 0.4 | V |
| M |
Może powstać sytuacja, w której pomimo oznak obecności znaczącej ilości wodorotlenków sodu i/lub potasu krzywa miareczkowania nie wykazuje wyraźnego punktu przegięcia. W takim wypadku należy powtórzyć oznaczenie w izopropanolu.
5.2. Miareczkowanie w izopropanolu
5.2.1. Odczynniki
5.2.1.1. Izopropanol
5.2.1.2. Standardowy 1.0 N roztwór wodny kwasu chlorowodorowego
5.2.1.3. 0.1 N roztwór kwasu chkorowodorowego w izopropanolu przygotowany bezpośrednio przed użyciem przez rozcieńczenie 1.0 N wodnego roztworu kwasu chlorowodorowego izopropanolem.
5.2.2. Aparatura
5.2.2.1. Zwykłe szklane wyposażenie laboratoryjne
5.2.2.2. Pehametr, korzystnie, jeśli z rejestratorem
5.2.2.3. Szklana elektroda membranowa
5.2.2.4. Standardowa kalomelowa elektroda odniesienia
5.2.3. Procedura
Odważyć dokładnie w zlewce o pojemności 150 ml próbkę o wielkości 0.5–1.0 g. Jeżeli w próbce znajduje się amoniak, dodać kilka ziaren porowatych, umieścić zlewkę w eksykatorze próżniowym, usuwać amoniak za pomocą pompy wodnej tak długo, aż zapach jego będzie niewyczuwalny (około trzech godzin). Dodać 100 ml izopropanolu, rozpuścić lub zdyspergować pozostałość i miareczkować 0.1 N roztworem kwasu chlorowodorowego w izopropanolu (5.2.1.3.), rejestrując zmiany pH (5.2.2.2.).
5.2.4. Obliczenia
Jak w punkcie 5.1.4. Pierwszy punkt przegięcia występuje przy odczytanej wartości pH około 9.
5.3. Powtarzalność
Dla wodorotlenku sodu lub potasu na poziomie 5% m/m wyrażonym jako wodorotlenek sodu różnica między wynikami dwóch oznaczeń wykonywanych równolegle na tej samej próbce nie powinna przekraczać wartości bezwzględnej 0.25%.
X. Oznaczanie dichlorometanu oraz 1,1,1-trichloroetanu
1. Cel i zakres stosowania
Metoda opisuje oznaczanie dichlorometanu (chlorku metylenu) i 1,1,1-trichloroetanu (metylochloroformu) we wszystkich kosmetykach mogących zawieracie rozpuszczalniki.
2. Definicja
Zawartość dichlorometanu i 1,1,1-trichloroetanu w próbce wyrobu oznaczana zgodnie z tą metodą jest wyrażona w procentach masowych.
3. Zasada
W metodzie używana jest chromatografia gazowa z chloroformem jako wewnętrznym standardem.
4. Odczynniki
Wszystkie odczynniki muszą być czystości analitycznej.
4.1. Chloroform (CHCl3)
4.2. Czterochlorek węgla (CCl4)
4.3. Dichlorometan (CH2CI2)
4.4. 1,1,1-Trichloroetan (CH3CCl3)
4.5. Aceton
4.6. Azot
5. Aparatura
5.1. Typowe wyposażenie laboratoryjne
5.2. Chromatograf gazowy wyposażony w detektor przewodnictwa termicznego
5.3. Butelka do przenoszenia 50 do 100 ml
5.4. Ciśnieniowa strzykawka gazowa, 25 lub 50 µl
6. Procedura
6.1. Próbka pod normalnym ciśnieniem: odważyć dokładnie próbkę w kolbie stożkowej zamykanej korkiem. Wprowadzić dokładnie zważoną ilość chloroformu (4.1) jako standard wewnętrzny równoważny przewidywanej ilości dichlorometanu i 1,1,1-trichloroetanu zawartych w próbce. Dokładnie wymieszać.
6.2. Próbka pod zwiększonym ciśnieniem: zastosować metodę pobierania próbek opisaną w rozdziale o pobieraniu próbek, ale z następującymi poprawkami:
6.2.1. Po przeniesieniu próbki do butelki do przenoszenia (5.3.) wprowadzić następnie do tej butelki pewną objętość chloroformu (4.1.) jako standard wewnętrzny równoważny przewidywanej ilości dichlorometanu i/lub 1,1,1-trichloroetanu zawartych w próbce. Dokładnie wymieszać. Przemyć martwą objętość zaworu 0.5 ml czterochlorku węgla (4.2.). Po wysuszeniu oznaczyć dokładnie dodaną masę standardu wewnętrznego jako różnicę.
6.2.2. Po napełnieniu strzykawki próbką dysza strzykawki powinna być przepłukana azotem (4.6.), tak aby żadna pozostałość próbki nie została w niej przed wprowadzeniem do chromatografu.
6.2.3. Po każdym pobraniu próbki powierzchnia zaworu i część przenosząca powinny być przepłukane kilkakrotnie acetonem (4.5.) (z użyciem strzykawki do iniekcji podskórnych) i wysuszone dokładnie azotem (4.6.).
6.2.4. Dla każdej analizy należy wykonać pomiary z użyciem dwóch różnych butelek do przenoszenia i powtarzać pięciokrotnie pomiary dla każdej butelki.
7. Warunki analizy chromatograficznej
7.1. Kolumna wstępna: połączenia rurowe: stal nierdzewna, długość: 300 mm, średnica: 3 lub 6 mm. Wypełnienie: to samo, które jest używane do wypełnienia kolumny analitycznej.
7.2. Kolumna
Faza stacjonarna jest przygotowana z Hallcomidu M18 osadzonego na chromosorbie. Kolumna powinna mieć zdolność rozdzielczą „R” równą lub wyższą niż 1,5
| R = 2 | d` (r2 – r1) |
| W1 + W2 |
oznaczono:
| r1 i r2 | = czasy retencji (w minutach) |
| W1 i W2 | = szerokość pików w połowie wysokości (w milimetrach) |
| d` | = szybkość przesuwu papieru (w milimetrach na minutę) |
7.3 Poniższe (przykładowe) kolumny spełniają wymagania metody:
7.4
| Kolumna | l | II |
| materiał: | rurka ze stali nierdzewnej | rurka ze stali nierdzewnej |
| długość: | 350 cm | 400 cm |
| średnica: | 3 mm | 6 mm |
| nośnik: |
|
|
| chromosorb: | WAW | WAW-DMCS-HP |
| analiza sitowa: | 100 do 120 mesh | 60 do 80 mesh |
| faza stacjonarna: | Hallcomid M18, 10% | Hallcomid M18, 20% |
Warunki temperaturowe mogą się różnić zależnie od aparatu. W przykładach ustalono je następująco:
| Kolumna: | l | II |
| temperatury: |
|
|
| kolumna: | 650C | 750C |
| dozownik: | 1500C | 1250C |
| detektor: | 1500C | 2000C |
| gaz nośny: |
|
|
| szybkość przepływu helu: | 45 ml/min | 60ml/min |
| ciśnienie wlotowe: | 2,5 bara | 2 bary |
| dozowanie: | 15 µl | 15 µl |
8. Mieszanina do ustalenia współczynnika korekcyjnego
Sporządzić następującą dokładnie odważoną mieszaninę w kolbie stożkowej z korkiem: dichlorometan (4.3.) 30% (m/m), 1,1,1-trichloroetan (4.4.), 35% (m/m), chloroform (4.1.), 35% (m/m).
9. Obliczenia
9.1. Obliczenie współczynnika korekcyjnego substancji „p” w stosunku do substancji „a” wybranej jako standard wewnętrzny
Podać najpierw substancję oznaczoną „p”, gdzie:
| kp | = jej współczynnik korekcyjny, |
| mp | = jej masa w mieszaninie, |
| Ap | = powierzchnia jej piku, |
Podać następnie substancję „a”, gdzie:
| ka | = jej współczynnik odpowiedzi (przyjęty za jedność), |
| Ma | = jej masa w mieszaninie, |
| Aa | = powierzchnie jej piku, |
| następnie | kp = | mp x Aa |
| Ma x Ap |
Jako przykłady otrzymano następujące współczynniki korekcyjne (dla chloroformu k = 1)
| Dichlorometan: | k1 = 0,78 ± 0,03 |
| 1,1,1-trichloroetan: | k2 = 1,00 ± 0,03 |
9.2. Obliczyć % (m/m) dichlorometanu i 1,1,1-trichloroetanu w analizowanej próbce:
Przyjęto:
ma = masa (w gramach) wprowadzonego chloroformu,
MS = masa (w gramach) analizowanej próbki,
Aa = powierzchnia piku chloroformu,
A1 = powierzchnia piku dichlorometanu,
A2 = powierzchnia piku 1,1,1-trichloroetanu,
następnie:
| % (m/m) CH2CI2 = | ma x A1 x k1 x 100 |
| Aa x MS |
| % (m/m) CH3CCI3 = | ma x A2 x k2 x 100 |
| Aa x MS |
XI. Oznaczanie rezorcyny w szamponach i płynach do włosów
1. Cel i zakres
Metoda podaje opis oznaczania rezorcyny metodą chromatografii gazowej w szamponach i płynach do włosów. Metoda jest odpowiednia dla stężeń od 0,1 do 2,0% masowych w próbce.
2. Definicja
Zawartość rezorcyny w próbce określona tą metodą jest wyrażana jako procent masowy.
3. Zasada
Rezorcyna i 3,5-dihydroksytoluen (5-metylorezorcyna) dodawany jako standard wewnętrzny są wydzielone z próbki metodą chromatografii cienkowarstwowej. Oba związki są izolowane przez zdrapywanie ich plam z płytki cienkowarstwowej i ekstrahowanie metanolem. Na zakończenie ekstrahowane związki suszy się, sililuje i oznacza metodą chromatografii gazowej.
4. Odczynniki
Wszystkie odczynniki muszą być czystości analitycznej
4.1. Kwas solny 25% (m/m)
4.2. Metanol
4.3. Etanol 96% (v/v)
4.4. Gotowe płytki do chromatografii cienkowarstwowej (TLC) (pokryte żelem krzemionkowym, tworzywowe lub aluminiowe) ze wskaźnikiem fluorescencyjnym. Należy dezaktywować płytki w następujący sposób: spryskać zwykłe pokryte żelem krzemionkowym płytki wodą do chwili, aż staną się szkliste. Pozostawić spryskane płytki do wyschnięcia w temperaturze pokojowej na jedną do trzech godzin.
Uwaga. Jeśli płytki nie zostaną zdezaktywowane, mogą wystąpić straty rezorcyny na skutek nieodwracalnej adsorpcji na żelu krzemionkowym.
4.5. Roztwór rozwijający: aceton-chloroform-kwas octowy (20 : 75 : 5 obj.)
4.6. Standardowy roztwór rezorcyny: rozpuścić 400 mg rezorcyny w 100 ml 96% etanolu (4.3.) (1 ml odpowiada 4 000 ug rezorcyny).
4.7. Roztwór standardu wewnętrznego: rozpuścić 400 mg 3,5-dihydroksytoluenu (DHT) w 100 ml 96% etanolu (4.3.) 1 ml odpowiada 4 000 µg DHT).
4.8. Mieszanina standardowa: zmieszać 10 ml roztworu (4.6.) i 10 ml roztworu (4.7.) w 100 ml kolbie miarowej, uzupełnić 96% etanolem (4.3.) do kreski i wymieszać (1 ml odpowiada 400 µg rezorcyny i 400 µg DHT).
4.9. Środki sililujące:
4.9.1. N,O-bis-(trimetylosililo)trójfluoroacetamid (BSTFA)
4.9.2. Sześciometylodisilazan (HMDS)
4.9.3. Trimetylochlorosilan (TMCS)
5. Aparatura
5.1. Zwykłe wyposażenie do chromatografii cienkowarstwowej i gazowej
5.2. Szkło laboratoryjne
6. Procedura
6.1. Przygotowanie próbek
6.1.1. Zważyć dokładnie do 150 ml zlewki próbkę analityczną (m gramów) produktu, która zawiera około 20 do 50 mg rezorcyny.
6.1.2. Zakwaszać kwasem solnym (4.1.) dopóki mieszanina nie stanie się kwaśna (średnio potrzeba 2 do 4 ml), dodać 10 ml (40 mg DHT) roztworu standardu wewnętrznego (4.7.) i wymieszać. Przenieść z etanolem (4.3.) do 100 ml kolby miarowej, uzupełnić do kreski etanolem i wymieszać.
6.1.3. Nanieść 250 µl roztworu (6.1.2.) na zdezaktywowaną płytkę z żelem krzemionkowym (4.4.) jako ciągłą linię o długości około 8 cm. Należy zadbać o to, aby otrzymać linię tak wąską, jak jest to możliwe.
6.1.4. Nanieść 250 µl mieszaniny standardowej (4.8.) na tę samą płytkę w ten sam sposób (6.1.3.).
6.1.5. Nakroplić w dwóch punktach linii początkowej po 5 µl każdego z roztworów (4.6. i 4.7.) dla ułatwienia umiejscowienia po rozwinięciu płytki.
6.1.6. Rozwijać płytkę w komorze bez arkusza bibuły (niewysyconej) zawierającej rozpuszczalnik rozwijający (4.5.) do chwili, kiedy czoło rozpuszczalnika osiągnie linię w odległości 12 cm od linii początkowej; zwykle trwa to około 45 minut. Wysuszyć płytkę na powietrzu i umiejscowić strefę rezorcyny/DHT w świetle nadfioletowym o krótkich falach (254 nm). Te dwa związki mają zwykle te same wartości Rf. Oznaczyć pasma ołówkiem w odległości 2 mm od zewnętrznej linii brzegowej pasm. Usunąć te strefy z płytki i zebrać adsorbent z każdego pasma w kolbie 10 ml.
6.1.7. Ekstrahować adsorbent zawierający próbkę i adsorbent zawierający mieszaninę standardową, każdy w następujący sposób: dodać 2 ml metanolu (4.2.) i ekstrahować w ciągu jednej godziny z ciągłym mieszaniem. Przesączyć mieszaninę i powtórzyć ekstrakcję w ciągu dalszych 15 minut z 2 ml metanolu.
6.1.8. Połączyć ekstrakty i odparować rozpuszczalnik, susząc przez noc w eksykatorze próżniowym zawierającym odpowiedni środek suszący. Nie korzystać z żadnego ogrzewania.
6.1.9. Sililować pozostałość (6.1.8.), jak wskazano w punkcie 6.1.9.1. i 6.1.9.2.
6.1.9.1. Dodać 200 µl BSTFA (4.9.1.) mikrostrzykawką i pozostawić mieszaninę w zamkniętym naczyniu na 12 godzin w temperaturze pokojowej.
6.1.9.2. Dodać 200 µl HMDS (4.9.2.) i 100 µl TMCS (4.9.3.) mikrostrzykawką i ogrzewać mieszaninę w ciągu 30 minut w 600C w zamkniętym naczyniu. Schłodzić mieszaninę.
6.2. Chromatografia gazowa
6.2.1. Warunki chromatograficzne
Kolumna musi mieć zdolność rozdzielczą R równą lub lepszą niż 1,5, gdzie:
| R= | 2d ̀̀(r2 - r1) |
| w1 + w2 |
w którym:
| r1 i r2 | – czasy retencji w minutach dwóch pików, |
| w1 i w2 | – szerokości tych dwóch pików w połowie wysokości, w mm, |
| d ̀̀ | – szybkość przesuwu papieru w mm na minutę. |
Następujące kolumny i warunki chromatografii gazowej uznano za odpowiednie:
Kolumna – materiał: stal kwasoodporna, długość 200 cm, średnica wewnętrzna: ~ 3 mm, wypełnienie: 10% OV-17 na Chromosorbie WAW 100 do 120mesh.
Detektor płomieniowo-jonizacyjny – temperatury: kolumna 185°C (izotermiczna), detektor 250°C, dozownik 250°C, gaz nośny – azot, przepływ 45 ml/min.
Dla ustawienia przepływów wodoru i powietrza należy przestrzegać instrukcji producentów.
6.2.2. Wprowadzić strzykawką 1 do 3 µl roztworów otrzymanych w punkcie 6.1.9. do chromatografu gazowego. Wykonać pięć injekcji dla każdego roztworu (6.1.9.), zmierzyć powierzchnię pików, obliczyć średnią wartość i stosunek powierzchni pików: S = powierzchnia pików rezorcyny/po-wierzchnia piku DHT.
7. Obliczenia
Stężenie rezorcyny w próbce, wyrażone w procentach masowych (% m/m) jest wyrażone przez wzór:
| % (m/m) rezorcyny = | 4 | x | S próbki |
| M | S mieszaniny standartowej |
w którym:
| M | – próbka analityczna w gramach (6.1.1.) |
| S próbki | – średni stosunek powierzchni pików według 6.2.2. dla roztworu próbki |
| S mieszaniny standardowej | – średni stosunek powierzchni pików według 6.2.2. dla mieszaniny standardowej. |
8. Powtarzalność
Dla zawartości rezorcyny około 0,5% różnica między wynikami dwu równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć wartości absolutnej 0,025%.
XII. Oznaczanie metanolu w stosunku do etanolu lub propan-2-olu
1. Cel i zakres
Metoda opisuje analizę metanolu metodą chromatografii gazowej we wszystkich kosmetykach (łącznie z aerozolowymi). Oznaczać można względne poziomy stężeń od 0 do 10%.
2. Definicja
Zawartość metanolu oznaczana tą metodą wyrażana jest w procentach masowych metanolu w stosunku do etanolu lub propan-2-olu.
3. Zasada
Oznaczenie wykonuje się metodą chromatografii gazowej.
4. Odczynniki
Należy używać odczynników analitycznych.
4.1. Metanol
4.2. Absolutny etanol
4.3. Propan-2-ol
4.4. Chloroform, z którego usunięto alkohole przez płukanie wodą
5. Aparatura
5.1. Chromatograf gazowy z detektorem katarometrycznym dla aerozoli, z detektorem płomieniowo-jonizacyjnym dla próbek nieaerozolowych.
5.2. Kolby miarowe, 100 ml
5.3. Pipety, 2 ml, 20 ml, od 0 do 1 ml
5.4. Mikrostrzykawki 0 do 100 µl i 0 do 5 µl (tylko dla próbek aerozolowych), specjalne szczelne gazowe strzykawki z zaworem suwakowym.
6. Procedura
6.1. Przygotowanie próbek
6.1.1. Próbki wyrobów aerozolowych są przygotowywane i następnie analizowane metodą chromatografii gazowej w warunkach jak w punkcie 6.2.1.
6.1.2. Próbki wyrobów nieaerozolowych przygotowywane według wymienionego powyżej rozdziału II są rozcieńczane wodą do poziomu stężenia od 1 do 2% etanolu lub proan-2-olu i następnie analizowane metodą chromatografii gazowej w warunkach jak w punkcie 6.2.2.
6.2. Chromatografia gazowa
6.2.1. Dla próbek aerozolowych używany jest detektor katarometryczny
6.2.1.1. Wypełnienie kolumny: 10% Hallcomid M 18 na Chromosorbie WAW 100 do 200 mesh
6.2.1.2. Kolumna musi mieć zdolność rozdzielczą R równą lub lepszą niż 1,5, gdzie:
| R = | 2 | d ̀̀r2 – d ̀r1 |
|
| w1 + w2 | |||
w której:
| r1 i r2 | – czasy retencji w minutach dla dwóch pików |
| w1 i w2 | – szerokości tych dwóch pików w połowie wysokości |
| d ̀̀ | – szybkość przesuwu papieru w mm na minutę |
6.2.1.3. Następujące warunki pozwalają na uzyskanie tego rozdziału:
Kolumna: materiał – stal kwasoodporna, długość 3–5 metrów, średnica – 3 mm,
Natężenie prądu mostka katarometru – 150 mA,
Gaz nośny – hel, ciśnienie – 2,5 bara, przepływ – 45 ml/min,
Temperatury: dozownik 150°C, detektor 150°C, piec kolumny 65°C.
Dokładność pomiarów powierzchni pików można poprawić poprzez zastosowanie integracji elektronicznej.
6.2.2. Dla próbek nieaerozolowych
6.2.2.1. Wypełnienie kolumny: Chromosorb 105 lub Porapak QS i używa się detektora płomieniowo-jonizacyjnego.
6.2.2.2. Kolumna musi mieć zdolność rozdzielczą R równą lub lepszą niż 1,5, gdzie:
| R = | 2 | d ̀̀r2 – d ̀r1 |
|
| w1 + w2 | |||
| r1 i r2 | – czasy retencji w minutach dla dwóch pików |
| w1 i w2 | – szerokości tych dwóch pików w połowie wysokości |
| d ̀̀ | – szybkość przesuwu papieru w mm na minutę |
6.2.2.3. Rozdział uzyskano w następujących warunkach:
Kolumna; materiał – stal kwasoodporna, długość – 2 metry, średnica – 3 mm,
Czułość elektrometru: 8 x 10-10A,
Gaz nośny: azot, ciśnienie – 2,1 bara, przepływ – 40 ml/min,
Gaz pomocniczy: wodór, ciśnienie – 1,5 bara, przepływ – 20 ml/min,
Temperatury: dozownik – 150°C, detektor – 230°C, piec kolumny 120 do 130°C.
7. Standardowy wykres
7.1. Do wykonania analizy metodą chromatografii gazowej według 6.2.1. (kolumna Hallcomid M 18) należy używać poniższych mieszanin standardowych. Mieszaniny przygotowuje się przez odmierzenie pipetami, jednak należy ustalić dokładną ilość składnika przez bezpośrednie zważenie pipety lub kolby po każdym dodaniu składnika.
| Stężenie względne | Metanol | Etanol lub propan-2-ol (ml) | Woda dodana do objętości (ml) |
| W przybliżeniu 2,5 | 0,5 | 20 | 100 |
| W przybliżeniu 5,0 | 1,0 | 20 | 100 |
| W przybliżeniu 7,5 | 1,5 | 20 | 100 |
| W przybliżeniu 10,0 | 2,0 | 20 | 100 |
Wprowadzić 2 do 3 µl do chromatografu, stosując warunki podane w punkcie 6.2.1. Obliczyć stosunek powierzchni pików (metanol/etanol) lub (metanol/propan-2-ol) dla wszystkich mieszanin. Wykreślić standardowy wykres z użyciem oznaczeń:
OŚ X: % metanolu w stosunku do etanolu lub propan-2-olu,
OŚ Y: stosunek powierzchni pików (metanol/etanol) lub (metanol/propan-2-ol).
7.2. Dla wykonania analizy metodą chromatografii gazowej według punktu 6.2.2. (kolumna Porapak OS lub Chromosorb 105) należy używać poniższych mieszanin standardowych. Mieszaniny przygotowuje się przez odmierzanie mikrostrzykawką i pipetą, jednak należy ustalić dokładną ilość składnika przez bezpośrednie zważenie pipety lub kolby po każdym dodaniu składnika.
| Stężenie względne | Metanol (µl) | Etanol lub propan-2-ol (ml) | Woda dodana do objętości (ml) |
| W przybliżeniu 2,5 | 50 | 2 | 100 |
| W przybliżeniu 5,0 | 100 | 2 | 100 |
| W przybliżeniu 7,5 | 150 | 2 | 100 |
| W przybliżeniu 10,0 | 200 | 2 | 100 |
Wprowadzić 2 do 3 µl do chromatografu, stosując warunki podane w punkcie 6.2.2. Obliczyć stosunek powierzchni pików (metanol/etanol) lub (metanol/propan-2-ol) dla wszystkich mieszanin. Wykreślić standardowy wykres z użyciem oznaczeń:
OŚ X: % metanolu w stosunku do etanolu lub propan-2-olu,
OŚ Y: stosunek powierzchni pików (metanol/etanol) lub (metanol/propan-2-ol).
7.3. Standardowy wykres musi być linią prostą
8. POWTARZALNOŚĆ
Dla zawartości metanolu 5% w stosunku do etanolu lub propan-2-olu różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,25%.
- Data ogłoszenia: 2003-01-27
- Data wejścia w życie: 2003-02-11
- Data obowiązywania: 2004-10-07
- Dokument traci ważność: 2020-01-02

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA