
REKLAMA
Dziennik Ustaw - rok 2023 poz. 1938
USTAWA
z dnia 17 sierpnia 2023 r.
o zmianie ustawy o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych oraz niektórych innych ustaw1)
Art. 1. [Ustawa o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych]
1) w art. 1 po pkt 1 dodaje się pkt 1a w brzmieniu:
„1a) zakres tajemnicy refundacyjnej;”;
2) użyte w art. 1 w pkt 4, w art. 2 w pkt 4, w art. 9 w ust. 6, w art. 11 w ust. 2 w pkt 5, w ust. 5 w pkt 2-4 i w ust. 7 w pkt 3, w art. 13 w ust. 1 we wprowadzeniu do wyliczenia, w ust. 3-5 i w ust. 6b, w art. 18 w ust. 1 w pkt 1, w art. 24 w ust. 1 w pkt 1-3, w art. 26 w pkt 1 w lit. ba i w pkt 2 w lit. ba, w art. 27 w pkt 4 w lit. ba, w art. 37 w ust. 7 oraz w art. 39 w ust. 3e w pkt 3, w różnej liczbie i różnym przypadku, wyrazy „urzędowa cena zbytu” zastępuje się użytymi w odpowiedniej liczbie i odpowiednim przypadku wyrazami „cena zbytu netto”;
3) w art. 2:
a) pkt 5 otrzymuje brzmienie:
„5) cena hurtowa - cenę zbytu netto leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego powiększoną o urzędową marżę hurtową;”,
b) po pkt 5 dodaje się pkt 5a w brzmieniu:
„5a) cena hurtowa brutto - cenę hurtową powiększoną o należny podatek od towarów i usług;”,
c) w pkt 6 wyrazy „do podmiotów uprawnionych do obrotu” zastępuje się wyrazami „przez wnioskodawcę podmiotom uprawnionym”,
d) po pkt 11 dodaje się pkt 11a w brzmieniu:
„11a) lek wytwarzany na terytorium Rzeczypospolitej Polskiej - lek, w odniesieniu do którego każde działanie podejmowane przez przedsiębiorcę niebędącego osobą zagraniczną w rozumieniu art. 3 pkt 5 ustawy z dnia 6 marca 2018 r. o zasadach uczestnictwa przedsiębiorców zagranicznych i innych osób zagranicznych w obrocie gospodarczym na terytorium Rzeczypospolitej Polskiej (Dz. U. z 2022 r. poz. 470) i prowadzące do jego powstania, w tym zakup i przyjmowanie w miejscu wytwarzania przez wytwórcę materiałów używanych do produkcji oraz produkcja, odbywa się w całości na terytorium Rzeczypospolitej Polskiej, z wyłączeniem zakupu leku, jego pakowania lub przepakowywania, magazynowania i dystrybucji, a także czynności kontrolnych związanych z tymi działaniami, w tym zwalnianiem do obrotu;”,
e) po pkt 17 dodaje się pkt 17a i 17b w brzmieniu:
„17a) prezentacja - lek, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny tego samego wnioskodawcy i posiadający tę samą nazwę, w przypadku którego pomijane są: dawka, wielkość opakowania, smak i kolor;
17b) produkt leczniczy terapii zaawansowanej - produkt leczniczy terapii zaawansowanej w rozumieniu art. 2 ust. 1 lit. a rozporządzenia (WE) nr 1394/2007 Parlamentu Europejskiego i Rady z dnia 13 listopada 2007 r. w sprawie produktów leczniczych terapii zaawansowanej i zmieniającego dyrektywę 2001/83/WE oraz rozporządzenie (WE) nr 726/2004 (Dz. Urz. UE L 324 z 10.12.2007, str. 121, z późn. zm.2));”,
f) po pkt 20a dodaje się pkt 20b w brzmieniu:
„20b) substancja czynna wytwarzana na terytorium Rzeczypospolitej Polskiej - substancję czynną, w odniesieniu do której każde działanie podejmowane przez przedsiębiorcę niebędącego osobą zagraniczną w rozumieniu art. 3 pkt 5 ustawy z dnia 6 marca 2018 r. o zasadach uczestnictwa przedsiębiorców zagranicznych i innych osób zagranicznych w obrocie gospodarczym na terytorium Rzeczypospolitej Polskiej i prowadzące do jej powstania, w tym zakup i przyjmowanie w miejscu wytwarzania przez wytwórcę materiałów używanych do jej produkcji oraz produkcja, odbywa się w całości na terytorium Rzeczypospolitej Polskiej, z wyłączeniem dopuszczenia do kolejnych etapów wytwarzania substancji czynnej, w tym jej pakowania, przepakowywania, ponownego etykietowania, magazynowania oraz dystrybucji, a także czynności kontrolnych związanych z tymi działaniami;”,
g) pkt 26 otrzymuje brzmienie:
„26) urzędowa cena zbytu - cenę zbytu netto leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego powiększoną o należny podatek od towarów i usług;”;
4) w art. 3:
a) w ust. 1 po wyrazach „Funduszu” dodaje się wyrazy „zatwierdzonym w trybie, o którym mowa w art. 121 ust. 4 ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych, zwanej dalej „ustawą o świadczeniach”, albo ustalonym w trybie, o którym mowa w art. 121 ust. 5 tej ustawy, z uwzględnieniem jego późniejszych zmian, o których mowa w art. 124 ustawy o świadczeniach, oraz środków pochodzących z instrumentów dzielenia ryzyka, o których mowa w art. 11 ust. 5, a także zwrotów, o których mowa w art. 34 ust. 1-1b”,
b) w ust. 2 w pkt 1 w lit. a wyrazy „ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych, zwanej dalej „ustawą o świadczeniach” ” zastępuje się wyrazami „ustawy o świadczeniach”,
c) uchyla się ust. 4;
5) w art. 6:
a) w ust. 1 w pkt 4 po wyrazie „żywieniowego” dodaje się przecinek i wyrazy „wyrób medyczny”,
b) po ust. 2 dodaje się ust. 2a w brzmieniu:
„2a. Kwota odpłatności przez pacjenta, o której mowa w ust. 2, ulega obniżeniu o:
1) 10% - w przypadku gdy lek jest wytwarzany na terytorium Rzeczypospolitej Polskiej albo gdy do jego wytworzenia wykorzystano substancję czynną wytworzoną na terytorium Rzeczypospolitej Polskiej,
2) 15% - w przypadku gdy lek jest wytwarzany na terytorium Rzeczypospolitej Polskiej z wykorzystaniem substancji czynnej wytwarzanej na terytorium Rzeczypospolitej Polskiej
- przy czym koszt tej obniżki jest finansowany ze środków Funduszu.”,
c) po ust. 8 dodaje się ust. 8a-8e w brzmieniu:
„8a. Wysokość limitów finansowania za jednostkę surowca farmaceutycznego służącego do sporządzenia leku recepturowego wyznacza się na poziomie 15 percentyla rozkładu cen danego surowca farmaceutycznego w roku poprzedzającym na podstawie danych przekazywanych przez apteki do Funduszu, zgodnie z art. 45 ust. 2.
8b. Świadczeniobiorca, nabywając lek recepturowy sporządzony z surowców nabytych przez aptekę po cenie nabycia wyższej niż limit finansowania, oprócz odpłatności ryczałtowej, o której mowa w ust. 5, dopłaca różnicę w wysokości różnicy między ceną nabycia surowców farmaceutycznych służących do sporządzenia leku recepturowego, a wysokością limitu finansowania danych surowców farmaceutycznych.
8c. Limit finansowania surowca farmaceutycznego w każdym kolejnym roku, nie może być wyższy niż 30 percentyl rozkładu cen danego surowca farmaceutycznego z roku poprzedzającego rok ubiegły.
8d. Prezes Funduszu, ustala corocznie, nie później niż do dnia 1 czerwca danego roku, wykaz limitów finansowania, o których mowa w ust. 8a. Limity te obowiązują od dnia 1 lipca danego roku.
8e. Wykaz, o którym mowa w ust. 8d, Fundusz podaje do publicznej wiadomości na swojej stronie internetowej oraz w Biuletynie Informacji Publicznej Funduszu.”;
6) w art. 7:
a) ust. 1 i 2 otrzymują brzmienie:
„1. Ustala się urzędową marżę hurtową w wysokości 6% ceny zbytu netto leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, nie niższą niż 50 groszy i nie wyższą niż:
1) 150 zł - w przypadku, o którym mowa w art. 6 ust. 1 pkt 1;
2) 2000 zł - w przypadkach, o których mowa w art. 6 ust. 1 pkt 2 i 3.
2. Przedsiębiorcy prowadzący obrót hurtowy w rozumieniu art. 72 ust. 3 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne są obowiązani stosować cenę zbytu netto wraz z marżą, o której mowa w ust. 1, oraz właściwą stawkę podatku od towarów i usług niezależnie od miejsca dostawy leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego będącego przedmiotem tego obrotu.”,
b) ust. 4 otrzymuje brzmienie:
„4. Ustala się urzędową marżę detaliczną naliczaną od ceny hurtowej brutto leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo wyrobu medycznego stanowiącego podstawę limitu w danej grupie limitowej, w wysokości:
| od | do | zasada marży |
| - | 10,00 zł | 0,45 zł + 35% * x |
| 10,01 zł | 20,00 zł | 3,95 zł + 25% * (x - 10,00 zł) |
| 20,01 zł | 40,00 zł | 6,45 zł + 15% * (x - 20,00 zł) |
| 40,01 zł | 80,00 zł | 9,45 zł + 10% * (x - 40,00 zł) |
| 80,01 zł | 160,00 zł | 13,45 zł + 5% * (x - 80,00 zł) |
| 160,01 zł | 640,00 zł | 17,45 zł + 2,75% * (x - 160,00 zł) |
| 640,01 zł | | 30,65 zł + 2% * (x - 640,00 zł) |
- gdzie x oznacza cenę hurtową brutto leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo wyrobu medycznego stanowiącego podstawę limitu, uwzględniającą liczbę DDD leku, jednostek środka spożywczego specjalnego przeznaczenia żywieniowego w opakowaniu albo liczbę jednostkowych wyrobów medycznych, albo liczbę jednostek wyrobu medycznego.”,
c) w ust. 6 po wyrazach „o którym mowa w” dodaje się wyrazy „art. 43 ust. 1 pkt 2 i”,
d) ust. 8 otrzymuje brzmienie:
„8. Dla leków, o których mowa w art. 6 ust. 5, ustala się marżę detaliczną w wysokości 25% liczoną od kosztu jego sporządzenia, wynoszącą nie więcej niż koszt wykonania leku recepturowego przygotowywanego w warunkach aseptycznych, określony w przepisach wydanych na podstawie art. 6 ust. 10.”;
7) art. 8 otrzymuje brzmienie:
„Art. 8. Urzędowe ceny zbytu są zawarte w obwieszczeniach, o których mowa w art. 37 ust. 1 i 4. Urzędowe ceny zbytu, a także urzędowe marże hurtowe i detaliczne, mają charakter cen i marż sztywnych.”;
8) w art. 9:
a) ust. 2 otrzymuje brzmienie:
„2. Świadczeniodawca jest obowiązany nabywać leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne, dla których ustalono kategorie dostępności refundacyjnej, o których mowa w art. 6 ust. 1 pkt 2 i 3, po cenie nie wyższej niż urzędowa cena zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego:
1) stanowiącego podstawę limitu, w przypadku leku zawierającego tę samą substancję czynną co lek wyznaczający podstawę limitu, środka spożywczego specjalnego przeznaczenia żywieniowego albo wyrobu medycznego, jednak nie wyższą niż urzędowa cena zbytu danego leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo wyrobu medycznego, albo
2) będącego najtańszym odpowiednikiem nabywanego leku, w przypadku gdy podstawę limitu w danej grupie limitowej wyznacza lek z inną substancją czynną, jednak nie wyższą niż urzędowa cena zbytu danego leku
- uwzględniając liczbę DDD leku, jednostek środka spożywczego specjalnego przeznaczenia żywieniowego w opakowaniu albo liczbę jednostkowych wyrobów medycznych, albo liczbę jednostek wyrobu medycznego, powiększoną o marżę nie wyższą niż urzędowa marża hurtowa, a w przypadku nabywania od podmiotu innego niż przedsiębiorca prowadzący obrót hurtowy w rozumieniu art. 72 ust. 3 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne - po cenie nie wyższej niż urzędowa cena zbytu.”,
b) po ust. 2 dodaje się ust. 2a w brzmieniu:
„2a. Realizator programu polityki zdrowotnej w rozumieniu art. 5 pkt 29a ustawy o świadczeniach nabywa do tego programu leki, dla których ustalono kategorię dostępności refundacyjnej, o której mowa w art. 6 ust. 1 pkt 2, po cenie nie wyższej niż wynikająca z instrumentu dzielenia ryzyka określonego dla tego leku w decyzji administracyjnej albo urzędowej ceny zbytu leku stanowiącego podstawę limitu, uwzględniając liczbę DDD leku, powiększoną o marżę nie wyższą niż urzędowa marża hurtowa, a w przypadku nabywania od podmiotu innego niż przedsiębiorca prowadzący obrót hurtowy w rozumieniu art. 72 ust. 3 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne - po cenie nie wyższej niż urzędowa cena zbytu, w zależności od tego, która z tych wartości jest niższa.”,
c) w ust. 3 wprowadzenie do wyliczenia otrzymuje brzmienie:
„Dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, o których mowa w art. 6 ust. 1 pkt 4, cenę zbytu netto ustala się:”;
9) w art. 10 w ust. 3 uchyla się pkt 2;
10) w art. 11:
a) w ust. 1 po wyrazach „Objęcie refundacją” dodaje się wyrazy „albo odmowa objęcia refundacją”,
b) w ust. 2:
- wprowadzenie do wyliczenia otrzymuje brzmienie:
„Decyzja administracyjna o objęciu refundacją, o której mowa w ust. 1, zawiera:”,
- pkt 3 otrzymuje brzmienie:
„3) kategorię dostępności refundacyjnej;”,
- uchyla się pkt 8,
- po pkt 8 dodaje się pkt 9 w brzmieniu:
„9) zobowiązanie do dostarczenia wielkości dostaw w ujęciu rocznym, z uwzględnieniem podziału na poszczególne miesiące, jeżeli dotyczy, określonej wzorem, o którym mowa w art. 25 pkt 4, na dzień wydawania decyzji.”,
c) ust. 3 otrzymuje brzmienie:
„3. Decyzję administracyjną o objęciu refundacją, o której mowa w ust. 1, wydaje się na okres:
1) 3 lat - dla leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, dla których nieprzerwanie obowiązywała decyzja administracyjna o objęciu refundacją lub w stosunku do których decyzja dla odpowiednika refundowanego w ramach tej samej kategorii dostępności refundacyjnej i w tym samym wskazaniu obowiązywała nieprzerwanie, przez okres dłuższy niż 3 lata;
2) 2 lat - dla leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, dla których nieprzerwanie obowiązywała decyzja administracyjna o objęciu refundacją lub w stosunku do których decyzja dla odpowiednika refundowanego w ramach tej samej kategorii dostępności refundacyjnej i w tym samym wskazaniu obowiązywała nieprzerwanie, przez okres krótszy niż 3 lata, albo dla których wydawana jest pierwsza decyzja administracyjna o objęciu refundacją;
3) 3 lat albo 5 lat - dla leków spełniających kryteria, o których mowa w art. 13a ust. 2 pkt 1 lit. b.”,
d) po ust. 3 dodaje się ust. 3a w brzmieniu:
„3a. Okres obowiązywania decyzji administracyjnej o objęciu refundacją, o której mowa w ust. 1, nie może przekraczać terminu wygaśnięcia okresu wyłączności rynkowej, o którym mowa w art. 15 ust. 2 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, lub ochrony wynikającej z patentu, w zależności od tego, co nastąpi jako pierwsze.”,
e) ust. 4 otrzymuje brzmienie:
„4. Podwyższenie albo obniżenie ceny zbytu netto następuje w drodze zmiany decyzji administracyjnej o objęciu refundacją, o której mowa w ust. 1.”,
f) w ust. 5 po pkt 4 dodaje się pkt 4a w brzmieniu:
„4a) ustalenia zasad realizacji dostaw i ich rozliczeń dotyczących leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych stosowanych w ramach programu lekowego lub leków stosowanych w ramach chemioterapii;”,
g) po ust. 5 dodaje się ust. 5a w brzmieniu:
„5a. W przypadku gdy decyzja administracyjna o objęciu refundacją, o której mowa w ust. 1, zawiera instrument dzielenia ryzyka, o którym mowa w ust. 2 pkt 7, może on być rozliczany w sposób określony w tym instrumencie, nawet jeżeli decyzja ta wygasła albo została uchylona.”,
h) ust. 6 otrzymuje brzmienie:
„6. Minister właściwy do spraw zdrowia, w drodze decyzji administracyjnej, ustala cenę zbytu netto leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, o którym mowa w art. 6 ust. 1 pkt 4.”,
i) w ust. 7 w pkt 3 kropkę zastępuję się średnikiem i dodaje się pkt 4 w brzmieniu:
„4) instrumenty dzielenia ryzyka, jeżeli zostały ustalone.”,
j) ust. 9 otrzymuje brzmienie:
„9. Decyzja, o której mowa w ust. 6, wygasa w dniu umieszczenia leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego w obwieszczeniu, o którym mowa w art. 37 ust. 1. Wygaśnięcie stwierdza minister właściwy do spraw zdrowia w drodze decyzji administracyjnej.”,
k) w ust. 11 pkt 2 otrzymuje brzmienie:
„2) cena zbytu netto leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, o którym mowa w art. 6 ust. 1 pkt 4, została ustalona z urzędu.”,
l) dodaje się ust. 12 w brzmieniu:
„12. W przypadku decyzji administracyjnych, o których mowa w ust. 1 i 6, zawierających instrument dzielenia ryzyka oraz innych decyzji dotyczących tego instrumentu minister właściwy do spraw zdrowia doręcza kopie tych decyzji Prezesowi Funduszu oraz Prezesowi Agencji.”;
11) w art. 12:
a) we wprowadzeniu do wyliczenia skreśla się wyrazy „i ustaleniu urzędowej ceny zbytu”,
b) w pkt 13 na końcu dodaje się przecinek i dodaje się pkt 14 w brzmieniu:
„14) zobowiązania do zapewnienia ciągłości dostaw, o którym mowa w art. 25 pkt 4”;
12) w art. 13:
a) ust. 2 otrzymuje brzmienie:
„2. W pierwszej decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego wydanej po upływie okresu, o którym mowa w art. 11 ust. 3a, cena zbytu netto nie może być określona na poziomie wyższym niż 75% ceny zbytu netto określonej w ostatniej decyzji administracyjnej o objęciu refundacją tego leku, środka lub wyrobu wydanej przed upływem tego okresu.”,
b) po ust. 2 dodaje się ust. 2a i 2b w brzmieniu:
„2a. W przypadku gdy w ostatniej decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego wydanej przed upływem okresu, o którym mowa w art. 11 ust. 3a, był zawarty instrument dzielenia ryzyka obniżający cenę zbytu netto (cena efektywna), mechanizm, o którym mowa w ust. 2, stosuje się w odniesieniu do:
1) instrumentu dzielenia ryzyka, o którym mowa w art. 11 ust. 5, obniżającego cenę efektywną na poziomie nie wyższym niż 75% ceny efektywnej określonej w ostatniej decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego albo
2) ceny zbytu netto oraz ceny efektywnej określonej w taki sposób, aby suma obniżek takich cen wynosiła łącznie co najmniej 25 punktów procentowych względem takich cen określonych w ostatniej decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego.
2b. W przypadku gdy w ostatniej decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego wydanej przed upływem okresu, o którym mowa w art. 11 ust. 3a, nie był zawarty instrument dzielenia ryzyka, spełnienie warunku, o którym mowa w ust. 2, może być również zrealizowane przez obniżenie ceny do poziomu 75% ceny zbytu netto określonej w ostatniej decyzji administracyjnej w instrumencie dzielenia ryzyka.”,
c) ust. 6 i 6a otrzymują brzmienie:
„6. Cena zbytu netto, o której mowa w ust. 5, z uwzględnieniem liczby DDD w opakowaniu jednostkowym, nie może być wyższa niż:
1) 75% ceny zbytu netto prezentacji o najniższym koszcie za DDD, liczonym według ceny zbytu netto jedynego odpowiednika refundowanego w danym wskazaniu;
2) cena zbytu netto:
a) odpowiednika wyznaczającego podstawę limitu albo
b) najtańszego odpowiednika, o ile podstawę limitu w danej grupie limitowej wyznacza lek niebędący odpowiednikiem leku, który jest przedmiotem wniosku
- w przypadku kolejnego odpowiednika refundowanego w danym wskazaniu.
6a. Cena zbytu netto ustalona w decyzji administracyjnej o objęciu refundacją, o której mowa w art. 11 ust. 1, nie może być wyższa niż cena zbytu netto leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, obowiązująca w dniu złożenia wniosku, o którym mowa w art. 24 ust. 1 pkt 1, 1a lub 1b, w odniesieniu do leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który w dniu złożenia tego wniosku był zawarty w wykazie, o którym mowa w art. 37 ust. 1.”,
d) po ust. 6a dodaje się ust. 6aa w brzmieniu:
„6aa. W przypadku gdy cena zbytu netto w przeliczeniu na jedno opakowanie lub na jednego pacjenta ulega obniżeniu na skutek zawarcia instrumentu dzielenia ryzyka, w kolejnej decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego cena efektywna, o której mowa w ust. 2a, nie może być wyższa niż w decyzji dotychczasowej.”,
e) w ust. 8:
- wprowadzenie do wyliczenia otrzymuje brzmienie:
„Minister właściwy do spraw zdrowia ustala cenę zbytu netto leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, o którym mowa w art. 6 ust. 1 pkt 4, przy uwzględnieniu kryteriów:”,
- pkt 4 otrzymuje brzmienie:
„4) ceny zbytu netto:
a) leków posiadających tę samą nazwę międzynarodową albo inne nazwy międzynarodowe, ale podobne działanie terapeutyczne,
b) środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, przy zastosowaniu następujących kryteriów:
- tych samych lub zbliżonych wskazań lub przeznaczeń,
- podobnej skuteczności”,
- w części wspólnej po wyrazie „żywieniowego” dodaje się przecinek i wyrazy „wyrobami medycznymi”;
13) po art. 13 dodaje się art. 13a w brzmieniu:
„Art. 13a. 1. Wnioskodawca, składając wniosek, o którym mowa w art. 24 ust. 1 pkt 1, dla leku wytwarzanego na terytorium Rzeczypospolitej Polskiej albo leku wytworzonego z wykorzystaniem substancji czynnej wytworzonej na terytorium Rzeczypospolitej Polskiej, może wnioskować o przyznanie maksymalnie jednej z preferencji administracyjnych i jednej z preferencji ekonomicznych, o których mowa w ust. 2, a w przypadku wytwarzania leku na terytorium Rzeczypospolitej Polskiej z wykorzystaniem substancji czynnej wytworzonej na terytorium Rzeczypospolitej Polskiej - po dwie takie preferencje.
2. Preferencje, o których mowa w ust. 1, dotyczą:
1) w zakresie preferencji administracyjnych:
a) zwolnienia z obowiązku zapłaty kwoty przekroczenia w przypadku przekroczenia całkowitego budżetu na refundację, o którym mowa w art. 4 - w takim przypadku koszt ten ponosi Fundusz,
b) wydania pierwszej decyzji administracyjnej o objęciu refundacją na okres 3 lat, a każdej kolejnej decyzji administracyjnej o objęciu refundacją na okres 5 lat, pod warunkiem że proponowana cena zbytu netto nie przekroczy 150% ceny zbytu netto leku stanowiącej podstawę limitu w grupie limitowej albo 150% kosztu DDD najtańszego odpowiednika, jeżeli lek nie stanowi odpowiednika leku stanowiącego podstawę limitu w grupie limitowej, w której znajduje się lek, chyba że lek nie był dotychczas wytwarzany na terytorium Rzeczypospolitej Polskiej - wówczas pierwsza decyzja administracyjna o objęciu refundacją jest wydawana na okres 5 lat,
c) zwolnienia z negocjacji cenowych z Komisją Ekonomiczną, o której mowa w art. 17, leku o kategorii dostępności refundacyjnej, o której mowa w art. 6 ust. 1 pkt 1,
d) rozpatrzenia wniosku w terminie nie dłuższym niż 90 dni - dla leków o kategorii dostępności refundacyjnej, o której mowa w art. 6 ust. 1 pkt 1, oraz nie dłuższym niż 120 dni - dla leków refundowanych stosowanych w ramach chemioterapii lub programów lekowych;
2) w zakresie preferencji ekonomicznych:
a) obniżenia opłaty za złożenie wniosku lub jego uzupełnienie, o których mowa w art. 32 ust. 1, do poziomu 50%,
b) obniżenia do 50% opłaty, o której mowa w art. 35 ust. 3,
c) ustalenia ceny zbytu netto w wysokości 75% ceny zbytu netto jedynego odpowiednika refundowanego w danym wskazaniu - w przypadku, o którym mowa w art. 13 ust. 6 pkt 1.
3. Do wniosku, o którym mowa w art. 24 ust. 1 pkt 1, wnioskodawca, o którym mowa w ust. 1, dołącza dokument potwierdzający wytwarzanie leku na terytorium Rzeczypospolitej Polskiej lub wytwarzanie leku z wykorzystaniem substancji czynnej wytworzonej na terytorium Rzeczypospolitej Polskiej.
4. Minister właściwy do spraw zdrowia w terminie 30 dni od dnia otrzymania wniosku, o którym mowa w ust. 1, wydaje postanowienie, o którym mowa w art. 123 ustawy z dnia 14 czerwca 1960 r. - Kodeks postępowania administracyjnego (Dz. U. z 2023 r. poz. 775 i 803), określające przyznane wnioskodawcy preferencje, biorąc pod uwagę działania prowadzące do powstania leku wytwarzanego na terytorium Rzeczypospolitej Polskiej.”;
14) w art. 14:
a) w ust. 1 we wprowadzeniu do wyliczenia po wyrazie „odpłatności” dodaje się wyrazy „na dzień wydania decyzji”,
b) w ust. 2 w zdaniu drugim wyrazy „miesięcznej terapii” zastępuje się wyrazami „30-dniowej terapii”;
15) w art. 15:
a) ust. 1-3 otrzymują brzmienie:
„1. Minister właściwy do spraw zdrowia ustala grupy leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, w ramach których wyznacza się podstawę limitu. Grup limitowych nie tworzy się w odniesieniu do leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, o których mowa w art. 6 ust. 1 pkt 4.
2. Do grupy limitowej kwalifikuje się:
1) lek posiadający tę samą nazwę międzynarodową albo inne nazwy międzynarodowe, ale podobne działanie terapeutyczne i zbliżony mechanizm działania;
2) środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny, przy zastosowaniu następujących kryteriów:
a) tych samych lub zbliżonych wskazań lub przeznaczeń, w których są refundowane,
b) podobnej skuteczności.
3. Po zasięgnięciu opinii Prezesa Agencji, której podstawą jest porównanie wielkości kosztów uzyskiwania podobnego efektu zdrowotnego lub dodatkowego efektu zdrowotnego w danym stanie klinicznym, dopuszcza się w trakcie obowiązywania decyzji refundacyjnej tworzenie odrębnych lub wspólnych grup limitowych oraz dokonanie zmiany w grupach limitowych, która następuje przez zmianę z urzędu decyzji administracyjnej o objęciu refundacją.”,
b) po ust. 3 dodaje się ust. 3a i 3b w brzmieniu:
„3a. Decyzja, o której mowa w ust. 3, podlega natychmiastowemu wykonaniu.
3b. Skutek prawny zmian w grupach limitowych następuje w obwieszczeniu, o którym mowa w art. 37 ust. 1, opublikowanym po dacie wydania decyzji, o której mowa w ust. 3.”,
c) ust. 4 i 5 otrzymują brzmienie:
„4. Podstawę limitu w danej grupie limitowej dla leków, o których mowa w art. 6 ust. 1 pkt 1, stanowi cena hurtowa za DDD leku o najwyższej spośród najniższych cen zbytu netto za DDD, który dopełnia 25% obrotu ilościowego liczonego według DDD, zrealizowanego w tej grupie limitowej w miesiącu poprzedzającym o 3 miesiące ogłoszenie obwieszczenia, o którym mowa w art. 37 ust. 1.
5. Podstawę limitu w przypadku:
1) środka spożywczego specjalnego przeznaczenia żywieniowego - stanowi cena hurtowa za jednostkę tego środka, dla którego najwyższy spośród najniższych kosztów 30-dniowego stosowania według cen zbytu netto dopełnia 25% obrotu ilościowego zrealizowanego w tej grupie limitowej w miesiącu poprzedzającym o 3 miesiące ogłoszenie obwieszczenia, o którym mowa w art. 37 ust. 1;
2) wyrobu medycznego - stanowi cena hurtowa za jednostkę tego wyrobu medycznego, dla którego najwyższa spośród najniższych cen zbytu netto za jednostkę tego wyrobu medycznego dopełnia 25% obrotu ilościowego zrealizowanego w tej grupie limitowej w miesiącu poprzedzającym o 3 miesiące ogłoszenie obwieszczenia, o którym mowa w art. 37 ust. 1.”,
d) po ust. 7 dodaje się ust. 7a w brzmieniu:
„7a. W przypadku gdy w danej grupie limitowej więcej niż jeden lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny jest pierwszym odpowiednikiem, do obliczeń, o których mowa w ust. 7, wykorzystuje się cenę hurtową produktu odpowiednio o najniższej cenie zbytu netto za DDD lub o najniższym koszcie 30-dniowego stosowania według cen zbytu netto, lub o najniższej cenie zbytu netto za jednostkę.”,
e) ust. 11 otrzymuje brzmienie:
„11. Podstawę limitu w danej grupie limitowej leków, o których mowa w art. 6 ust. 1 pkt 2 i 3, stanowi najwyższa cena hurtowa leku, którego cena zbytu netto za DDD nie przekracza średniej ceny zbytu netto za DDD leków w tej grupie limitowej w roku poprzedzającym rok ustalenia podstawy. Przepis ust. 7 stosuje się odpowiednio.”,
f) po ust. 11 dodaje się ust. 11a w brzmieniu:
„11a. Podstawę limitu nowo tworzonej grupy limitowej leków, o których mowa w art. 6 ust. 1 pkt 2 i 3, stanowi cena hurtowa za DDD leku, który według deklaracji złożonej we wniosku o objęcie refundacją dopełnia 100% szacowanego zapotrzebowania na te leki.”,
g) w ust. 13 we wprowadzeniu do wyliczenia wyrazy „ust. 11 i 12” zastępuje się wyrazami „ust. 11-12”,
h) ust. 14 otrzymuje brzmienie:
„14. W przypadku gdy DDD jest inna niż najczęściej stosowana dobowa dawka leku (PDD), minister właściwy do spraw zdrowia może wyznaczyć podstawę limitu na podstawie PDD.”;
16) po art. 16 dodaje się art. 16a i art. 16b w brzmieniu:
„Art. 16a. 1. Minister właściwy do spraw zdrowia tworzy opisy programów lekowych po zasięgnięciu opinii konsultanta krajowego lub konsultanta wojewódzkiego z odpowiedniej dziedziny medycyny, a w uzasadnionych przypadkach również medycznego towarzystwa naukowego z danej dziedziny medycyny. Opis programu lekowego podlega opiniowaniu przez Prezesa Agencji.
2. Minister właściwy do spraw zdrowia przedstawia opis programu lekowego do zaopiniowania wnioskodawcy po wydaniu przez Prezesa Agencji opinii, o której mowa w ust. 1. Wnioskodawca jest obowiązany przedstawić opinię w terminie nie dłuższym niż 7 dni od dnia otrzymania opisu programu lekowego. Opinia wnioskodawcy nie jest wiążąca dla ministra właściwego do spraw zdrowia.
3. Opis programu lekowego może przewidywać powołanie przez Prezesa Funduszu zespołu koordynacyjnego dla danego programu lekowego.
4. Zmiany opisu programu lekowego dokonuje minister właściwy do spraw zdrowia.
5. W przypadku dokonywania zmiany, o której mowa w ust. 4, minister właściwy do spraw zdrowia może zasięgnąć opinii konsultanta krajowego lub konsultanta wojewódzkiego z odpowiedniej dziedziny medycyny oraz Prezesa Agencji, a w uzasadnionych przypadkach również medycznego towarzystwa naukowego z danej dziedziny medycyny.
Art. 16b. 1. W przypadku gdy opis programu lekowego tak stanowi, Prezes Funduszu powołuje zespół koordynacyjny dla tego programu.
2. Do zadań zespołu koordynacyjnego należy:
1) kwalifikacja świadczeniobiorców do programu lekowego;
2) ocena skuteczności terapii w trakcie trwania programu lekowego;
3) rozstrzyganie o wyłączeniu świadczeniobiorców z programu lekowego;
4) składanie półrocznych raportów do Prezesa Funduszu z realizacji danego programu lekowego;
5) składanie do ministra właściwego do spraw zdrowia propozycji zmian, kontynuacji lub zaprzestania finansowania programu lekowego na podstawie danych przekazywanych przez świadczeniodawców realizujących dany program lekowy do Funduszu i zespołu koordynacyjnego, w szczególności dotyczących oceny skuteczności terapii.
3. Członkowie zespołu koordynacyjnego są powoływani przez Prezesa Funduszu.
4. Do zespołu koordynacyjnego może zostać powołana osoba posiadająca tytuł specjalisty z dziedziny medycyny właściwej dla danego programu lekowego.
5. Członkowie zespołu koordynacyjnego wybierają ze swojego składu przewodniczącego, jego zastępców i sekretarza.
6. Zespół koordynacyjny obraduje na posiedzeniach, w tym z wykorzystaniem środków komunikacji elektronicznej umożliwiających porozumiewanie się na odległość.
7. W posiedzeniu zespołu koordynacyjnego mogą brać udział, bez prawa głosu:
1) przedstawiciel Prezesa Funduszu;
2) przedstawiciel ministra właściwego do spraw zdrowia.
8. Prezes Funduszu określi, w drodze zarządzenia, regulamin zespołu koordynacyjnego określający jego organizację, szczegółowe zadania, sposób i tryb działania oraz uczestnictwo w pracach tego zespołu.
9. Obsługę administracyjną oraz prawną zespołu koordynacyjnego i jego członków w zakresie spraw prowadzonych przez ten zespół zapewnia wskazany przez Prezesa Funduszu, w porozumieniu z ministrem właściwym do spraw zdrowia, świadczeniodawca realizujący umowy o udzielanie świadczeń opieki zdrowotnej w zakresie programów lekowych.
10. Prezes Funduszu odwołuje członka zespołu koordynacyjnego z własnej inicjatywy albo na umotywowany wniosek ministra właściwego do spraw zdrowia w przypadku:
1) rezygnacji ze stanowiska;
2) choroby trwale uniemożliwiającej wykonywanie powierzonych zadań;
3) uchylania się od wykonywania obowiązków członka zespołu koordynacyjnego albo nieprawidłowego wykonywania tych obowiązków.”;
17) w art. 18:
a) w ust. 1 uchyla się pkt 2,
b) w ust. 2 uchyla się pkt 1 i 3,
c) ust. 3 otrzymuje brzmienie:
„3. Komisja na podstawie dokumentu stanowiącego wynik negocjacji, sporządzonego w postaci elektronicznej, podpisanego przez strony negocjacji, podejmuje uchwałę w drodze głosowania elektronicznego za pomocą Systemu Obsługi List Refundacyjnych, o którym mowa w art. 30a ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia, zwanego dalej „SOLR”, i przekazuje ją niezwłocznie ministrowi właściwemu do spraw zdrowia. Przepis art. 24 ust. 6c stosuje się odpowiednio.”;
18) po art. 18 dodaje się art. 18a w brzmieniu:
„Art. 18a. 1. Po podjęciu przez Komisję uchwały, o której mowa w art. 18 ust. 3, wnioskodawca nie może zmienić treści wniosku, o którym mowa w art. 24 ust. 1, w szczególności w zakresie ceny zbytu netto i instrumentu dzielenia ryzyka, o którym mowa w art. 11 ust. 2 pkt 7.
2. Minister właściwy do spraw zdrowia może, po podjęciu przez Komisję uchwały, o której mowa w art. 18 ust. 3, przeprowadzić dodatkowe negocjacje z wnioskodawcą w zakresie, o którym mowa w art. 18 ust. 1 pkt 1, 3 i 4, biorąc pod uwagę kryteria, o których mowa w art. 19 ust. 2, jeżeli uzna to za konieczne ze względu na niezaspokojone potrzeby świadczeniobiorców oraz możliwości płatnicze podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych.”;
19) w art. 19:
a) ust. 1 otrzymuje brzmienie:
„1. Rozpatrując wnioski, o których mowa w art. 24 ust. 1 pkt 1-2, 4 i 5, Komisja prowadzi negocjacje w składzie trzyosobowym, zwanym dalej „zespołem negocjacyjnym”, z tym że w każdym składzie powinien znaleźć się przedstawiciel Prezesa Funduszu.”,
b) po ust. 1 dodaje się ust. 1a-1c w brzmieniu:
„1a. Spośród członków zespołu negocjacyjnego przewodniczący Komisji wskazuje przewodniczącego zespołu negocjacyjnego.
1b. Przewodniczący Komisji może dopuścić większą liczbę członków Komisji do udziału w negocjacjach. 1c. Wnioskodawca podczas negocjacji jest reprezentowany przez nie więcej niż 3 osoby. Reprezentantem wnioskodawcy nie może być konsultant krajowy lub konsultant wojewódzki.”;
20) w art. 20:
a) w ust. 1 pkt 2 otrzymuje brzmienie:
„2) być członkami organów spółek handlowych lub przedstawicielami przedsiębiorców prowadzących działalność gospodarczą w zakresie świadczenia usług prawnych, marketingowych lub doradczych dotyczących wytwarzania, obrotu lub refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego;”,
b) ust. 2 otrzymuje brzmienie:
„2. Kandydaci na członków oraz członkowie Komisji składają ministrowi właściwemu do spraw zdrowia oświadczenie o niezachodzeniu okoliczności określonych w ust. 1, pod rygorem odpowiedzialności karnej za składanie fałszywych oświadczeń z art. 233 § 6 ustawy z dnia 6 czerwca 1997 r. - Kodeks karny (Dz. U. z 2022 r. poz. 1138, z późn. zm.3)), zwane dalej „deklaracją o braku konfliktu interesów”, odpowiednio przed powołaniem oraz przed każdym posiedzeniem Komisji, obejmujące okres 3 lat poprzedzających złożenie oświadczenia. Składający deklarację jest obowiązany do zawarcia w niej klauzuli o następującej treści: „Jestem świadomy odpowiedzialności karnej za złożenie fałszywego oświadczenia.”. Klauzula ta zastępuje pouczenie organu o odpowiedzialności karnej za składanie fałszywych oświadczeń.”,
c) w ust. 5:
- po pkt 1 dodaje się pkt 1a w brzmieniu:
„1a) numer PESEL osoby składającej oświadczenie;”,
- po pkt 2 dodaje się pkt 2a w brzmieniu:
„2a) numery PESEL osób pozostających we wspólnym pożyciu, o których mowa w pkt 2;”;
21) w art. 22 ust. 2 otrzymuje brzmienie:
„2. Członkom Komisji przysługuje wynagrodzenie za udział w posiedzeniach Komisji lub negocjacjach w wysokości nieprzekraczającej 3500 zł za udział w każdym posiedzeniu Komisji lub każdych negocjacjach, jednak miesięcznie nie więcej niż trzykrotność przeciętnego miesięcznego wynagrodzenia w gospodarce narodowej w poprzednim roku kalendarzowym, ogłaszanego corocznie przez Prezesa Głównego Urzędu Statystycznego, w drodze obwieszczenia, w Dzienniku Urzędowym Rzeczypospolitej Polskiej „Monitor Polski”.”;
22) w art. 24:
a) w ust. 1 pkt 4 otrzymuje brzmienie:
„4) ustalenie albo zmianę ceny zbytu netto leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo wyrobu medycznego, o którym mowa w art. 6 ust. 1 pkt 4;”,
b) po ust. 1 dodaje się ust. 1a w brzmieniu:
„1a. W przypadku wniosku, o którym mowa w art. 24 ust. 1 pkt 2, który został złożony w okresie obowiązywania decyzji administracyjnej o objęciu refundacją trwającą przed wygaśnięciem okresu wyłączności rynkowej, o którym mowa w art. 15 ust. 2 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, lub ochrony wynikającej z patentu, oraz w okresie 12 miesięcy obowiązywania pierwszej decyzji administracyjnej o objęciu refundacją wydanej po zaistnieniu tych okoliczności, wniosek taki nie wywołuje skutku prawnego i pozostawia się go bez rozpoznania.”,
c) w ust. 2:
- po pkt 1 dodaje się pkt 1a w brzmieniu:
„1a) informację, w jakich wskazaniach jest refundowany ten produkt w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA);”,
- w pkt 5 kropkę zastępuje się średnikiem i dodaje się pkt 6 w brzmieniu:
„6) dokument patentowy lub dokument, z którego wynika udzielenie dodatkowego okresu wyłączności rynkowej, jeżeli dotyczy.”,
d) po ust. 2 dodaje się ust. 2a w brzmieniu:
„2a. W przypadku gdy informacje dołączone zgodnie z ust. 2 uległy zmianie po dniu złożenia wniosku, o którym mowa w ust. 1, wnioskodawca jest obowiązany niezwłocznie, nie później niż do upływu terminu wyznaczonego w zawiadomieniu o zakończeniu postępowania oraz możliwości wypowiedzenia się co do dowodów i materiałów zebranych w sprawie, wynikającego z art. 10 ustawy z dnia 14 czerwca 1960 r. - Kodeks postępowania administracyjnego, do ich aktualizacji.”,
e) w ust. 4 wyrazy „o których mowa w art. 26 pkt 1 lit. h oraz i lub pkt 2 lit. h-j” zastępuje się wyrazami „o których mowa w art. 25 pkt 14 lit. c oraz art. 26 pkt 1 lit. h lub pkt 2 lit. h oraz i”,
f) po ust. 6a dodaje się ust. 6aa i 6ab w brzmieniu:
„6aa. Składający wniosek oświadcza pod rygorem odpowiedzialności karnej za składanie fałszywych oświadczeń z art. 233 § 6 ustawy z dnia 6 czerwca 1997 r. - Kodeks karny, że dane zawarte we wniosku oraz informacje i dokumenty, o których mowa w ust. 2, są zgodne z prawdą. Składający deklarację jest obowiązany dołączyć do wniosku klauzulę o następującej treści: „Jestem świadomy odpowiedzialności karnej za złożenie fałszywego oświadczenia.”. Klauzula ta zastępuje pouczenie organu o odpowiedzialności karnej za składanie fałszywych oświadczeń.
6ab. Oświadczenie, o którym mowa w ust. 6aa, nie dotyczy dokumentów, o których mowa w art. 25 pkt 14 lit. a oraz c, art. 25a pkt 14 lit. a-c, art. 25b pkt 14, art. 26 pkt 1 lit. h, pkt 2 lit. h oraz i, art. 28 pkt 7 lit. a i art. 29 pkt 5.”,
g) po ust. 6d dodaje się ust. 6e w brzmieniu:
„6e. Decyzje, postanowienia oraz inne pisma wydane w toku postępowania w sprawie refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego prowadzonego za pomocą SOLR uznaje się za doręczone z chwilą ich podpisania i umieszczenia w skrzynce odbiorcy.”,
h) w ust. 7 w pkt 2 wyrazy „oraz art. 26 pkt 2 lit. h-j” zastępuje się wyrazami „oraz art. 26 pkt 2 lit. h oraz i”;
23) skreśla się użyty w art. 25 w pkt 1, w art. 26 w pkt 1 w lit. a oraz w pkt 2 w lit. a, w art. 27 w pkt 1, w art. 28 w pkt 1, w art. 29 w pkt 1, w art. 32a w ust. 2 oraz w art. 37b w ust. 3 w pkt 1 wyraz „telefaks” i przecinek;
24) w art. 25:
a) pkt 3 i 4 otrzymują brzmienie:
„3) dowód dostępności w obrocie leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego w chwili składania wniosku, a w przypadku produktu leczniczego terapii zaawansowanej - zobowiązanie do zapewnienia gotowości technologicznej do jego wytworzenia na dzień składania wniosku;
4) zobowiązanie do zapewnienia ciągłości dostaw wraz z określeniem rocznej wielkości dostaw podanej w ujęciu miesięcznym - w przypadku objęcia refundacją; minimalna roczna wielkość dostaw dla leku będącego jedynym produktem refundowanym w danym wskazaniu wynosi nie mniej niż 110% szacowanej rocznej populacji, a dla produktu, w przypadku którego jest refundowany przynajmniej jeden odpowiednik w danym wskazaniu, nie może być niższa niż wartość określona wzorem:
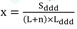
gdzie poszczególne symbole oznaczają:
x - minimalną deklarowaną roczną liczbę opakowań wnioskowanego leku,
Sddd - ilość zrefundowanych DDD substancji w danej dawce w grupie limitowej w ubiegłym roku,
L - liczbę leków w danej dawce w grupie limitowej na dzień składania wniosku,
n - liczbę leków w danej dawce, o których refundację wnioskuje wnioskodawca,
Lddd - liczbę DDD w opakowaniu wnioskowanego leku;”,
b) po pkt 5 dodaje się pkt 5a w brzmieniu:
„5a) deklarację zgodności UE, o której mowa w art. 19 rozporządzenia 2017/745 albo art. 17 rozporządzenia 2017/746, wzór oznakowania wyrobów medycznych oraz kopię certyfikatu jednostki notyfikowanej, o którym mowa w art. 56 rozporządzenia 2017/745 albo art. 51 rozporządzenia 2017/746, jeżeli dotyczy - w przypadku wyrobów medycznych;”,
c) po pkt 8 dodaje się pkt 8a w brzmieniu:
„8a) wskazanie obowiązującej stawki podatku od towarów i usług w rozumieniu ustawy z dnia 11 marca 2004 r. o podatku od towarów i usług (Dz. U. z 2023 r. poz. 1570, 1598 i 1852) dla wnioskowanego leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego;”,
d) w pkt 14 w lit. c uchyla się tiret czwarte;
25) w art. 25c:
a) w ust. 1 po wyrazach „w danym wskazaniu” dodaje się wyrazy „(wniosek kontynuacyjny)”,
b) dodaje się ust. 3 w brzmieniu:
„3. Jeżeli po złożeniu wniosku kontynuacyjnego, o którym mowa w ust. 1, następuje wygaśnięcie dotychczasowej decyzji dotyczącej refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, minister właściwy do spraw zdrowia umarza postępowanie wszczęte na podstawie tego wniosku.”;
26) w art. 26:
a) w pkt 1:
- uchyla się lit. i,
- w lit. m średnik zastępuje się przecinkiem i dodaje się lit. n w brzmieniu:
„n) dowód wzrostu kosztów związanych z produkcją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego oraz analizę ekonomiczną kosztów wytwarzania i działalności wytwórcy uzasadniającą wnioskowane podwyższenie ceny zbytu netto;”,
b) w pkt 2:
- uchyla się lit. j,
- w lit. n kropkę zastępuje się przecinkiem i dodaje się lit. o w brzmieniu:
„o) dowód wzrostu kosztów związanych z produkcją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego oraz analizę ekonomiczną kosztów wytwarzania i działalności wytwórcy uzasadniającą wnioskowane podwyższenie ceny zbytu netto.”;
27) w art. 28:
a) w pkt 4 wprowadzenie do wyliczenia otrzymuje brzmienie:
„dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny w danej wielkości lub dawce, jeżeli dotyczy:”,
b) w pkt 7 uchyla się lit. b;
28) w art. 30 dodaje się ust. 3 w brzmieniu:
„3. Wymagania, o których mowa w art. 25 pkt 14, nie dotyczą wniosków, o których mowa w art. 24 ust. 1 pkt 1, dla leku zawierającego substancję czynną, który nie ma odpowiednika refundowanego w danym wskazaniu, a który to lek albo jego odpowiednik był zawarty w wykazie, o którym mowa w art. 37 ust. 1, w danym wskazaniu, i otrzymał pozytywną rekomendację Prezesa Agencji. Przepis ust. 2 zdanie drugie stosuje się.”;
29) po art. 30 dodaje się art. 30a i art. 30b w brzmieniu:
„Art. 30a. 1. Minister właściwy do spraw zdrowia może wydać decyzję o objęciu refundacją i ustaleniu urzędowej ceny zbytu dla leku, w tym leku o kategorii dostępności OTC, który wymaga stosowania dłużej niż 30 dni w określonym stanie klinicznym i jest rekomendowany w wytycznych postępowania klinicznego, w przypadku gdy wnioskodawca nie złożył dotychczas wniosku o objęcie refundacją i ustalenie urzędowej ceny zbytu dla danego leku w danym wskazaniu oraz upłynęła dla tego leku wyłączność rynkowa.
2. Minister właściwy do spraw zdrowia zleca Prezesowi Agencji przygotowanie wykazu leków, o których mowa w ust. 1. Prezes Agencji przygotowuje wykaz, uwzględniając wiedzę z zakresu oceny technologii medycznych, w szczególności oczekiwane efekty zdrowotne, biorąc pod uwagę siłę interwencji, jakość danych naukowych, niezaspokojoną potrzebę zdrowotną, wielkość populacji docelowej oraz priorytety zdrowotne i wpływ na system ochrony zdrowia, w tym wpływ na budżet płatnika.
3. Agencja przekazuje ministrowi właściwemu do spraw zdrowia i publikuje w Biuletynie Informacji Publicznej wykaz, o którym mowa w ust. 2, w terminie 60 dni od daty otrzymania zlecenia.
4. Agencja ustala zasady tworzenia wykazu, o którym mowa w ust. 2, uwzględniając wiedzę z zakresu oceny technologii medycznych, w szczególności oczekiwane efekty zdrowotne, biorąc pod uwagę między innymi siłę interwencji, jakość danych naukowych, niezaspokojoną potrzebę zdrowotną, wielkość populacji docelowej oraz priorytety zdrowotne.
5. Minister właściwy do spraw zdrowia, po zasięgnięciu opinii Rady Przejrzystości, konsultantów krajowych z danej dziedziny medycyny oraz Rzecznika Praw Pacjenta, publikuje na swojej stronie internetowej oraz w Biuletynie Informacji Publicznej listę leków na podstawie wykazu, o którym mowa w ust. 2, przekazanego przez Agencję. Minister właściwy do spraw zdrowia informuje podmioty odpowiedzialne, których leki znajdują się na tej liście, o możliwości złożenia wniosku, o którym mowa w art. 24 ust. 1 pkt 1.
6. Do wniosków, o których mowa w ust. 5, nie stosuje się przepisów art. 35.
Art. 30b. 1. Tajemnicą refundacyjną są objęte informacje:
1) zawarte we wniosku, o którym mowa w art. 24 ust. 1, oraz w innych dokumentach składanych przez wnioskodawców lub wytworzonych w postępowaniu wszczętym takim wnioskiem;
2) z posiedzenia Komisji, o których mowa w art. 18 ust. 3;
3) dotyczące negocjacji, o których mowa w art. 19 ust. 1, oraz
4) uzyskane w trakcie prowadzonych negocjacji dodatkowych, o których mowa w art. 18a ust. 2.
2. Do przestrzegania tajemnicy refundacyjnej są obowiązani:
1) minister właściwy do spraw zdrowia;
2) osoby zatrudnione w urzędzie obsługującym ministra właściwego do spraw zdrowia;
3) Prezes Agencji, osoby zatrudnione w Agencji oraz członkowie Rady Przejrzystości;
4) Prezes Funduszu oraz osoby zatrudnione w Funduszu;
5) osoby odbywające staż, praktykę zawodową lub studencką w urzędach wymienionych w pkt 2-4 oraz osoby działające na zlecenie tych urzędów lub ministra właściwego do spraw zdrowia;
6) członkowie Komisji;
7) osoby zatrudnione w jednostce podległej ministrowi właściwemu do spraw zdrowia właściwej w zakresie systemów informacyjnych ochrony zdrowia, o której mowa w ustawie z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia, posiadające dostęp do SOLR i jej podwykonawcy.
3. Zachowanie tajemnicy refundacyjnej obowiązuje również po ustaniu zatrudnienia lub powołania, zakończeniu stażu lub praktyki, wykonaniu zlecenia przez osobę działającą na zlecenie lub podwykonawcę lub po ustaniu członkostwa w Komisji.
4. Minister właściwy do spraw zdrowia oraz osoby zatrudnione w urzędzie obsługującym tego ministra są zwolnieni z obowiązku przestrzegania tajemnicy refundacyjnej, jeżeli ujawnienie informacji objętych tą tajemnicą jest niezbędne do:
1) wykonywania zadań związanych z refundacją leków, środków spożywczych specjalnego przeznaczenia żywieniowego lub wyrobów medycznych przez Fundusz, Agencję lub świadczeniodawcę;
2) realizacji postanowień umów międzynarodowych, bilateralnych lub porozumień w zakresie wymiany informacji dotyczących refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego lub wyrobów medycznych;
3) wykonania obowiązków informacyjnych wobec Komisji Europejskiej.
5. W przypadku przekazania informacji objętych tajemnicą przedsiębiorstwa w rozumieniu art. 11 ust. 2 ustawy z dnia 16 kwietnia 1993 r. o zwalczaniu nieuczciwej konkurencji (Dz. U. z 2022 r. poz. 1233) innym organom krajowym, zagranicznym organom regulacyjnym albo innym podmiotom, z wyłączeniem sądów lub prokuratury, informacje i dokumenty przekazuje się wraz z zastrzeżeniem i pod warunkiem przestrzegania tej tajemnicy.”;
30) w art. 31:
a) po ust. 3 dodaje się ust. 3a i 3b w brzmieniu:
„3a. Do postępowań wszczętych na podstawie wniosku, o którym mowa w art. 24 ust. 1, nie stosuje się przepisów art. 98 ustawy z dnia 14 czerwca 1960 r. - Kodeks postępowania administracyjnego.
3b. Minister właściwy do spraw zdrowia może z urzędu, w drodze postanowienia, zawiesić postępowanie wszczęte na podstawie wniosku, o którym mowa w art. 24 ust. 1, na okres nie dłuższy niż 90 dni, jeżeli wymaga tego interes społeczny. Przepisu art. 102 ustawy z dnia 14 czerwca 1960 r. - Kodeks postępowania administracyjnego nie stosuje się.”,
b) w ust. 6 wyrazy „90 dni” zastępuje się wyrazami „120 dni”,
c) po ust. 7 dodaje się ust. 7a-7c w brzmieniu:
„7a. Obniżenie ceny zbytu netto na podstawie wniosku, o którym mowa w art. 24 ust. 1 pkt 3, złożonego nie później niż na 45 dni przed ogłoszeniem pierwszego po złożeniu tego wniosku obwieszczenia, o którym mowa w art. 37 ust. 1, obowiązuje od dnia, na który ustalono wykaz w tym obwieszczeniu. W przypadku złożenia wniosku po tym terminie obniżenie ceny zbytu netto nastąpi od dnia obowiązywania kolejnego wykazu.
7b. W przypadku gdy wniosek, o którym mowa w art. 24 ust. 1 pkt 3, dotyczy leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego objętego refundacją w więcej niż jednym wskazaniu lub w zakresie wskazań do stosowania lub dawkowania, lub sposobu podawania odmiennych niż określone w Charakterystyce Produktu Leczniczego, o której mowa w art. 10 ust. 2 pkt 11 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, jest traktowany jako wniosek dotyczący wniosku o obniżenie ceny zbytu netto dla wszystkich wskazań produktu o tym samym numerze GTIN zgodnym z systemem GS1 lub innym kodem jednoznacznie identyfikującym środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny znajdujących się w obwieszczeniu, o którym mowa w art. 37 ust. 1.
7c. Wniosek, o którym mowa w art. 24 ust. 1 pkt 5, albo wniosek o ponowne rozpatrzenie sprawy dotyczący tego wniosku rozpatruje się w terminie 60 dni, z tym że w przypadku konieczności uzupełnienia danych niezbędnych do rozpatrzenia wniosku, bieg tego terminu ulega zawieszeniu do dnia otrzymania uzupełnienia danych albo do dnia upływu terminu uzupełnienia wniosku.”,
d) uchyla się ust. 10-12;
31) w art. 32:
a) ust. 2 otrzymuje brzmienie:
„2. Opłaty, o których mowa w ust. 1, stanowią dochód budżetu państwa i każda z nich nie może być wyższa niż 15 000 zł.”,
b) po ust. 2 dodaje się ust. 2a w brzmieniu:
„2a. Opłata, o której mowa w ust. 1, za złożenie wniosku dotyczącego leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, którego cena zbytu netto wynosi co najmniej 10 000 zł lub którego koszt terapii rocznej pacjenta przekracza trzykrotność produktu krajowego brutto na jednego mieszkańca dla obszaru całej Rzeczypospolitej Polskiej, podaną w obwieszczeniu wydanym na podstawie art. 6 ustawy z dnia 26 października 2000 r. o sposobie obliczania wartości rocznego produktu krajowego brutto, nie może być wyższa niż 30 000 zł.”,
c) ust. 3 otrzymuje brzmienie:
„3. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, wysokość opłat, o których mowa w ust. 1, mając na uwadze rodzaj wniosku, stopień jego skomplikowania, nakład pracy i koszty związane z jego rozpatrywaniem.”;
32) w art. 32a w ust. 3 w pkt 3 wyrazy „opłaty za założenie konta” zastępuje się wyrazami „opłaty za złożenie wniosku o założenie konta”;
33) w art. 33:
a) w ust. 1:
- wprowadzenie do wyliczenia otrzymuje brzmienie:
„Minister właściwy do spraw zdrowia może uchylić decyzję administracyjną o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, w przypadku:”,
- pkt 4 otrzymuje brzmienie:
„4) gdy zobowiązanie, o którym mowa w art. 34 ust. 2, nie zostanie dotrzymane w zakresie dotyczącym zapewnienia ciągłości dostaw;”,
- dodaje się pkt 5-7 w brzmieniu:
„5) gdy zobowiązanie, o którym mowa w art. 11 ust. 2 pkt 9, nie zostanie dotrzymane w zakresie dotyczącym rocznej wielkości dostaw i nastąpi niezaspokojenie potrzeb świadczeniobiorców;
6) gdy wnioskodawca przestał spełniać warunki, o których mowa w art. 13a ust. 1;
7) gdy jednostka notyfikująca wycofa certyfikat zgodności, o którym mowa w art. 56 rozporządzenia 2017/745 albo art. 51 rozporządzenia 2017/746, dla wyrobu medycznego.”,
b) po ust. 1 dodaje się ust. 1a w brzmieniu:
„1a. Minister właściwy do spraw zdrowia uchyla decyzję administracyjną o objęciu refundacją leku, wydaną na podstawie art. 40 ust. 1, w przypadku wygaśnięcia, uchylenia lub skrócenia okresu obowiązywania decyzji administracyjnej wydanej przy uwzględnieniu kryteriów, o których mowa w art. 12, dotyczącej tego leku.”,
c) dodaje się ust. 4 w brzmieniu:
„4. Decyzja uchylająca decyzję administracyjną o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego podlega natychmiastowemu wykonaniu.”;
34) po art. 33 dodaje się art. 33a w brzmieniu:
„Art. 33a. Minister właściwy do spraw zdrowia z urzędu zmienia decyzję administracyjną o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, w przypadku gdy na podstawie przepisów innych ustaw ulegną zmianie elementy objęte tą decyzją.”;
35) w art. 34:
a) ust. 1 otrzymuje brzmienie:
„1. W przypadku gdy zobowiązanie, o którym mowa w art. 11 ust. 2 pkt 9, w zakresie dotyczącym rocznej wielkości dostaw lub ciągłości dostaw nie zostanie dotrzymane, wnioskodawca, który otrzymał decyzję administracyjną o objęciu refundacją, jest obowiązany do zwrotu do Funduszu kwoty stanowiącej iloczyn liczby niedostarczonych jednostkowych opakowań leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo jednostkowych wyrobów medycznych i ich urzędowej ceny zbytu, chyba że niewykonywanie tego zobowiązania jest następstwem działania siły wyższej albo wnioskodawca dopełnił obowiązku określonego w ust. 2.”,
b) po ust. 1 dodaje się ust. 1a i 1b w brzmieniu:
„1a. Niezależnie od zobowiązania, o którym mowa w ust. 1, w przypadku gdy zobowiązanie, o którym mowa w art. 11 ust. 2 pkt 9, w zakresie dotyczącym rocznej wielkości dostaw, z uwzględnieniem ujęcia miesięcznego, lub ciągłości dostaw nie zostanie dotrzymane, a świadczeniobiorca otrzyma lek sprowadzony w trybie określonym w art. 4 ust. 1, 8 lub 9 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, wnioskodawca, który otrzymał decyzję administracyjną o objęciu refundacją, jest obowiązany do zwrotu do Funduszu kwoty stanowiącej różnicę między ustaloną urzędową ceną zbytu dla tego leku a faktycznie poniesionymi przez Fundusz wydatkami na refundację tego leku.
1b. Przepis ust. 1a stosuje się również do środka spożywczego specjalnego przeznaczenia żywieniowego oraz wyrobu medycznego.”,
c) ust. 2 otrzymuje brzmienie:
„2. Przez niedotrzymanie zobowiązania dotyczącego ciągłości dostaw, o którym mowa w ust. 1, rozumie się brak obrotu hurtowego w rozumieniu art. 72 ust. 3 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, produktem objętym refundacją, polegający na dostarczeniu produktu do świadczeniodawcy lub hurtowni farmaceutycznych, zlokalizowanych na terytorium Rzeczypospolitej Polskiej, w okresie danego kwartału kalendarzowego w ilościach mniejszych niż wynikających ze zobowiązania do miesięcznych dostaw tego kwartału, określonych w decyzji administracyjnej o objęciu refundacją, lub zobowiązania, o którym mowa w art. 25 pkt 4, chyba że niewykonywanie tego zobowiązania jest następstwem działania siły wyższej, a w przypadku produktu leczniczego terapii zaawansowanej - niedotrzymanie zobowiązania do zapewnienia gotowości technologicznej do jego wytworzenia. Powyższego obowiązku nie stosuje się do leków będących przedmiotem przetargów centralnych prowadzonych przez Prezesa Funduszu w przypadku wyłonienia w tym przetargu dostawcy leków dla całej populacji chorych w danym wskazaniu refundacyjnym na okres co najmniej 12 miesięcy oraz do leków dostarczanych sezonowo.”,
d) po ust. 3 dodaje się ust. 3a-3d w brzmieniu:
„3a. Wnioskodawca, w przypadku leków, środków spożywczych specjalnego przeznaczenia żywieniowego lub wyrobów medycznych objętych refundacją w kategorii dostępności refundacyjnej, o której mowa w art. 6 ust. 1 pkt 1, zagrożonych brakiem dostępności na terytorium Rzeczypospolitej Polskiej, obowiązany jest dostarczać te produkty w równej części do co najmniej 10 przedsiębiorców prowadzących hurtownie farmaceutyczne, których zakres działalności, o którym mowa w art. 76 ust. 1 pkt 8 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, określony w załączniku do zezwolenia na prowadzenie hurtowni farmaceutycznej, nie zawiera ograniczeń asortymentu, na terenie Rzeczypospolitej Polskiej, z największym udziałem obrotu z aptekami ogólnodostępnymi pod względem liczby transakcji, w ilości koniecznej do zabezpieczenia pacjentów, o którym mowa w art. 36z ust. 1 tej ustawy.
3b. Wykaz hurtowni, o których mowa w ust. 3a, minister właściwy do spraw zdrowia ustala corocznie do dnia 30 kwietnia danego roku w oparciu o dane dotyczące obrotu refundowanymi lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego lub wyrobami medycznymi między hurtowniami farmaceutycznymi a aptekami ogólnodostępnymi za poprzedni rok kalendarzowy, zawarte w Zintegrowanym Systemie Monitorowania Obrotu Produktami Leczniczymi, o którym mowa w art. 72a ust. 1 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne.
3c. Minister właściwy do spraw zdrowia publikuje w Biuletynie Informacji Publicznej wykaz, o którym mowa w ust. 3b.
3d. Wykaz leków, środków spożywczych specjalnego przeznaczenia żywieniowego lub wyrobów medycznych, o których mowa w ust. 3a, minister właściwy do spraw zdrowia ustala w drodze obwieszczenia i ogłasza w Dzienniku Urzędowym ministra właściwego do spraw zdrowia, kierując się potrzebą zapewnienia możliwie równomiernego zabezpieczenia pacjentów.”,
e) uchyla się ust. 5,
f) ust. 6 otrzymuje brzmienie:
„6. Kwotę, o której mowa w ust. 1, ustala w drodze noty obciążeniowej Prezes Funduszu i podlega ona uiszczeniu w terminie 14 dni od dnia doręczenia tej noty.”;
36) po art. 34 dodaje się art. 34a w brzmieniu:
„Art. 34a. 1. Podmiot prowadzący aptekę, który posiada lek, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny objęty refundacją w kategorii dostępności refundacyjnej, o której mowa w art. 6 ust. 1 pkt 1, dla którego została wydana decyzja administracyjna, o której mowa w art. 24 ust. 1 pkt 5, albo dla którego wygasła decyzja administracyjna o objęciu refundacją, nabyty przed tymi zmianami, może w terminie 30 dni od daty obowiązywania wykazu ustalonego w nowym obwieszczeniu, o którym mowa w art. 37 ust. 1, zwrócić lek, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny podmiotowi, od którego nabył dany lek, środek lub wyrób, z żądaniem zwrotu zapłaconej ceny.
2. Przedsiębiorca prowadzący obrót hurtowy w rozumieniu art. 72 ust. 3 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, który posiada lek, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny, dla którego została wydana decyzja administracyjna, o której mowa w art. 24 ust. 1 pkt 5, albo dla którego wygasła decyzja administracyjna o objęciu refundacją, nabyty przed tymi zmianami, może w terminie 45 dni od daty obowiązywania wykazu ustalonego w nowym obwieszczeniu, o którym mowa w art. 37 ust. 1, zwrócić lek, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny podmiotowi, od którego nabył dany lek, środek lub wyrób, z żądaniem zwrotu zapłaconej ceny.”;
37) w art. 35:
a) ust. 2 otrzymuje brzmienie:
„2. W przypadku stwierdzenia, że analizy, o których mowa w ust. 1, nie spełniają wymagań określonych w przepisach wydanych na podstawie art. 24 ust. 7 pkt 2, Prezes Agencji wzywa wnioskodawcę do uzupełnienia wniosku za pomocą SOLR, wyznaczając mu termin nie krótszy niż 21 dni na uzupełnienie dokumentacji. Bieg terminów, o których mowa w ust. 8 oraz art. 31 ust. 4 i 5, ulega zawieszeniu do dnia uzupełnienia dokumentacji albo do dnia upływu wyznaczonego terminu na jej uzupełnienie. W przypadku braku należytego uzupełnienia wniosku we wskazanym terminie minister właściwy do spraw zdrowia umarza postępowanie w sprawie refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego.”,
b) po ust. 2 dodaje się ust. 2a i 2b w brzmieniu:
„2a. W przypadku gdy Prezes Agencji stwierdzi, że w związku z utworzeniem opisu programu lekowego przez ministra właściwego do spraw zdrowia na podstawie art. 16a ust. 1 i 2 nastąpiła zmiana populacji docelowej, Prezes Agencji wzywa wnioskodawcę do uzupełnienia analiz, o których mowa w ust. 1, za pomocą SOLR. Bieg terminów, o których mowa w ust. 8 oraz art. 31 ust. 4 i 5, ulega zawieszeniu do dnia uzupełnienia dokumentacji.
2b. W przypadku braku dostępu do SOLR na skutek awarii systemu lub działania siły wyższej termin na dokonanie czynności, o których mowa w ust. 2 i 2a, uznaje się za zachowany pod warunkiem dokonania w wyznaczonym terminie czynności w postaci papierowej. Dokumentacja złożona w postaci papierowej, po uzyskaniu dostępu do SOLR, jest wprowadzana przez Prezesa Agencji do systemu w terminie 3 dni roboczych od dnia przywrócenia funkcjonalności systemu.”,
c) po ust. 3 dodaje się ust. 3a-3c w brzmieniu:
„3a. Limit opłaty podlega corocznej waloryzacji o średnioroczny wskaźnik cen towarów i usług konsumpcyjnych ogółem ustalany na podstawie komunikatu Prezesa Głównego Urzędu Statystycznego, ogłaszanego w Dzienniku Urzędowym Rzeczypospolitej Polskiej „Monitor Polski”, na podstawie art. 94 ust. 1 pkt 1 lit. a ustawy z dnia 17 grudnia 1998 r. o emeryturach i rentach z Funduszu Ubezpieczeń Społecznych (Dz. U. z 2023 r. poz. 1251, 1429 i 1672).
3b. Waloryzacja następuje z początkiem miesiąca następującego po miesiącu, w którym opublikowano komunikat Prezesa Głównego Urzędu Statystycznego w sprawie przeciętnego wynagrodzenia w gospodarce narodowej, ogłoszony w Dzienniku Urzędowym Rzeczypospolitej Polskiej „Monitor Polski”, na podstawie art. 20 pkt 1 lit. a ustawy z dnia 17 grudnia 1998 r. o emeryturach i rentach z Funduszu Ubezpieczeń Społecznych. Limit zwaloryzowanej opłaty, o której mowa w ust. 3a, oblicza się w wysokości uwzględniającej jego rewaloryzację oraz wszystkie kolejne waloryzacje.
3c. Prezes Agencji publikuje zaktualizowaną wysokość opłaty w formie komunikatu w Biuletynie Informacji Publicznej Agencji.”,
d) po ust. 4 dodaje się ust. 4a i 4b w brzmieniu:
„4a. Prezes Agencji nie ujawnia informacji stanowiących tajemnicę przedsiębiorstwa w rozumieniu art. 11 ust. 2 ustawy z dnia 16 kwietnia 1993 r. o zwalczaniu nieuczciwej konkurencji, jeżeli wnioskodawca zastrzegł, że stanowią one tajemnicę przedsiębiorstwa.
4b. Wnioskodawca nie może zastrzec jako tajemnicy przedsiębiorstwa:
1) informacji dotyczących wskazań, w których lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny ma być refundowany;
2) treści projektu programu lekowego;
3) określenia grupy limitowej zgodnie z wnioskiem;
4) faktu przedstawienia we wniosku propozycji instrumentu dzielenia ryzyka, o którym mowa w art. 11 ust. 5;
5) informacji dotyczącej refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego w innych krajach;
6) opublikowanych informacji o skuteczności klinicznej i praktycznej;
7) informacji o bezpieczeństwie stosowania leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, z wyłączeniem informacji pochodzących z raportu okresowego o bezpieczeństwie (ang. Periodic Safety Update Report - PSUR);
8) informacji o stosunku kosztów do uzyskiwanych efektów zdrowotnych w wariancie bez instrumentu dzielenia ryzyka, o którym mowa w art. 11 ust. 5;
9) informacji o efektywności kosztowej ocenianego leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego;
10) informacji o wpływie na wydatki podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych i świadczeniobiorców w wariancie bez instrumentu dzielenia ryzyka, o którym mowa w art. 11 ust. 5.”,
e) po ust. 8 dodaje się ust. 8a-8c w brzmieniu:
„8a. W przypadku wydania przez Prezesa Agencji rekomendacji, z której wynika konieczność spełnienia dodatkowych warunków w celu objęcia refundacją danego leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, minister właściwy do spraw zdrowia zawiesza postępowanie w sprawie refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego do czasu uzupełnienia przez wnioskodawcę wniosku, o którym mowa w art. 24 ust. 1 pkt 1, o dokumentację potwierdzającą ich spełnienie.
8b. Zawieszenie postępowania, o którym mowa w ust. 8a, nie może trwać dłużej niż 2 miesiące, a w sprawach szczególnie skomplikowanych - 3 miesiące. W przypadku braku uzupełnienia wniosku w terminie, o którym mowa w zdaniu pierwszym, minister właściwy do spraw zdrowia umarza postępowanie w sprawie refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego.
8c. W przypadku uzupełnienia wniosku Prezes Agencji sprawdza w terminie 30 dni od dnia jego otrzymania, czy dokumentacja spełnia warunki, o których mowa w ust. 8a.”;
38) po art. 35 dodaje się art. 35a w brzmieniu:
„Art. 35a. 1. Analiza weryfikacyjna jest jawna.
2. W analizie weryfikacyjnej wyłączeniu jawności mogą podlegać wyłącznie informacje dotyczące instrumentów dzielenia ryzyka, o których mowa w art. 11 ust. 5, i kosztów z nich wynikających oraz niepublikowane wcześniej wyniki badań klinicznych, które wnioskodawca zastrzegł tajemnicą przedsiębiorstwa w rozumieniu art. 11 ust. 2 ustawy z dnia 16 kwietnia 1993 r. o zwalczaniu nieuczciwej konkurencji.”;
39) w art. 36:
a) ust. 1 otrzymuje brzmienie:
„1. Wnioski, o których mowa w art. 24 ust. 1 pkt 1-2, 4 i 5, minister właściwy do spraw zdrowia przekazuje Komisji za pomocą SOLR w celu przeprowadzenia negocjacji.”,
b) ust. 4 otrzymuje brzmienie:
„4. Negocjacje dla wniosków, o których mowa w art. 24 ust. 1 pkt 1-2, 4 i 5, mogą odbywać się w maksymalnie 3 turach, przy czym w przypadku wniosków, o których mowa w art. 24 ust. 1 pkt 1a i 1b, negocjacje nie mogą trwać dłużej niż 30 dni, licząc od dnia pierwszych negocjacji.”,
c) ust. 6 otrzymuje brzmienie:
„6. W przypadku braku porozumienia z wnioskodawcą Komisja podejmuje uchwałę, w której nie rekomenduje objęcia refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego. Uchwała Komisji przekazywana jest za pośrednictwem SOLR ministrowi właściwemu do spraw zdrowia, a wynik negocjacji wraz z protokołami negocjacyjnymi, z wyjątkiem informacji stanowiących tajemnicę przedsiębiorstwa w rozumieniu art. 11 ust. 2 ustawy z dnia 16 kwietnia 1993 r. o zwalczaniu nieuczciwej konkurencji, jest podawany do wiadomości publicznej na stronie podmiotowej Biuletynu Informacji Publicznej urzędu obsługującego tego ministra.”;
40) po art. 36 dodaje się art. 36a w brzmieniu:
„Art. 36a. 1. Analizy, o których mowa w art. 25 pkt 14 lit. c oraz art. 26 pkt 2 lit. h oraz i, są ważne przez jeden rok, licząc od dnia złożenia wniosku, ale nie dłużej niż przez 3 lata od dnia ich sporządzenia.
2. W przypadku gdy postępowanie wszczęte na podstawie wniosku, o którym mowa w art. 24 ust. 1, w odniesieniu do którego została wydana rekomendacja Prezesa Agencji, o której mowa w art. 35 ust. 6, nie zostanie zakończone w terminie jednego roku, licząc od daty złożenia tego wniosku, minister właściwy do spraw zdrowia umarza postępowanie.”;
41) w art. 37:
a) w ust. 2:
- po pkt 3 dodaje się pkt 3a i 3b w brzmieniu:
„3a) cenę zbytu netto;
3b) cenę hurtową brutto;”,
- uchyla się pkt 5,
b) ust. 2b otrzymuje brzmienie:
„2b. Leki i wyroby medyczne przysługujące świadczeniobiorcom, o których mowa w art. 43b ust. 1 ustawy o świadczeniach, minister właściwy do spraw zdrowia ogłasza w obwieszczeniu, o którym mowa w ust. 1.”,
c) po ust. 2b dodaje się ust. 2c w brzmieniu:
„2c. Minister właściwy do spraw zdrowia ogłasza w obwieszczeniu, o którym mowa w ust. 1, również:
1) leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne, o których mowa w art. 6 ust. 2a, oraz
2) technologie lekowe o wysokiej wartości klinicznej oraz technologie lekowe o wysokim poziomie innowacyjności.”,
d) ust. 4 i 5 otrzymują brzmienie:
„4. Minister właściwy do spraw zdrowia ogłasza, w drodze obwieszczenia, wykazy:
1) leków,
2) środków spożywczych specjalnego przeznaczenia żywieniowego,
3) wyrobów medycznych
- o których mowa w art. 6 ust. 1 pkt 4, w stosunku do których wydano ostateczną decyzję administracyjną o ustaleniu ceny zbytu netto.
5. Obwieszczenie, o którym mowa w ust. 4, zawiera:
1) dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny;
2) cenę zbytu netto;
3) urzędową cenę zbytu.”,
e) po ust. 5 dodaje się ust. 5a w brzmieniu:
„5a. W uzasadnionych przypadkach w obwieszczeniach, o których mowa w ust. 1 i 4, mogą zostać uwzględnione inne informacje niż określone w ust. 2 albo 5, dotyczące warunków objęcia refundacją i ustalenia ceny zbytu netto oraz dostępności leku, środka spożywczego specjalnego przeznaczenia żywieniowego oraz wyrobu medycznego, w zakresie:
1) zmiany danych odnoszących się do leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, w tym elementów ich identyfikacji wizualnej, lub danych wnioskodawcy, mogących powodować problemy z ich prawidłową identyfikacją przez osobę uprawnioną lub pacjenta;
2) zagrożeń związanych z dostępnością do leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, mogących mieć wpływ na istotne ograniczenie skali ich refundacji.”,
f) ust. 6 otrzymuje brzmienie:
„6. Obwieszczenia, o których mowa w ust. 1 i 4, są ogłaszane raz na 3 miesiące w dzienniku urzędowym ministra właściwego do spraw zdrowia co najmniej na 14 dni przed dniem, na który ustala się wykazy, o których mowa w ust. 1 i 4.”;
42) w art. 37b w ust. 4 po wyrazach „programów lekowych” dodaje się wyrazy „lub chemioterapii”;
43) w art. 40:
a) po ust. 1 dodaje się ust. 1a w brzmieniu:
„1a. Decyzję, o której mowa w ust. 1, minister właściwy do spraw zdrowia może wydać w przypadku wcześniejszego lub równoczesnego wydania decyzji administracyjnej o objęciu refundacją i ustaleniu ceny zbytu netto, wydanej przy uwzględnieniu kryteriów, o których mowa w art. 12, dotyczącej tego leku, na okres nie dłuższy niż decyzja o objęciu refundacją w zakresie wskazań do stosowania określonych w Charakterystyce Produktu Leczniczego, o której mowa w art. 10 ust. 2 pkt 11 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne.”,
b) ust. 2 otrzymuje brzmienie:
„2. Opinię, o której mowa w ust. 1, Rada Przejrzystości wydaje nie później niż w terminie 30 dni od dnia otrzymania zlecenia od ministra właściwego do spraw zdrowia, biorąc pod uwagę istotność stanu klinicznego, w którym ma być stosowany lek, skuteczność kliniczną i praktyczną, bezpieczeństwo stosowania, relację korzyści zdrowotnych do ryzyka stosowania, wpływ na wydatki podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych i świadczeniobiorców oraz istnienie alternatywnej technologii medycznej.”,
c) po ust. 3 dodaje się ust. 3a w brzmieniu:
„3a. Agencja przekazuje ministrowi właściwemu do spraw zdrowia nie później niż w terminie 90 dni przed upływem terminu obowiązywania opinii, o której mowa w ust. 1, informację o zbliżającym się upływie tego terminu.”;
44) w art. 40a ust. 1 otrzymuje brzmienie:
„1. Minister właściwy do spraw zdrowia może wydać decyzję administracyjną o objęciu refundacją dla technologii lekowej o wysokim poziomie innowacyjności, w przypadku gdy wnioskodawca nie złożył dotychczas wniosku o objęcie refundacją dla danego leku w tym wskazaniu.”;
45) w art. 43 w ust. 1 pkt 6 otrzymuje brzmienie:
„6) zwrotu refundacji ceny leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego łącznie z odsetkami ustawowymi za opóźnienie liczonymi od dnia, w którym wypłacono refundację, do dnia jej zwrotu, w terminie 14 dni od dnia otrzymania wezwania do zapłaty, jeżeli w wyniku weryfikacji lub kontroli informacji, o których mowa w pkt 2, lub kontroli, o której mowa w pkt 3 i 4, zostanie stwierdzone, że realizacja recepty nastąpiła z naruszeniem przepisów ustawy, ustawy o świadczeniach, ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, ustawy z dnia 5 grudnia 1996 r. o zawodach lekarza i lekarza dentysty (Dz. U. z 2023 r. poz. 1516, 1617 i 1831), ustawy z dnia 29 lipca 2005 r. o przeciwdziałaniu narkomanii lub ustawy z dnia 10 grudnia 2020 r. o zawodzie farmaceuty (Dz. U. z 2022 r. poz. 1873 oraz z 2023 r. poz. 1234);”;
46) skreśla się użyty w art. 44 w ust. 1 w zdaniu pierwszym i w ust. 2a oraz w art. 44a w ust. 1 wyraz „terapeutycznym”;
47) w art. 44:
a) uchyla się ust. 2d,
b) w ust. 3 wyrazy „Przepisy ust. 1-2d” zastępuje się wyrazami „Przepisy ust. 1-2c”;
48) w art. 44a:
a) uchyla się ust. 3,
b) w ust. 4 wyrazy „Przepisy art. 44 ust. 1-2d” zastępuje się wyrazami „Przepisy art. 44 ust. 1-2c”;
49) w art. 44b:
a) ust. 1 otrzymuje brzmienie:
„1. W przypadku recepty wystawionej dla świadczeniobiorcy w okresie ciąży albo połogu przez osoby wskazane w art. 43b ust. 3 ustawy o świadczeniach osoba wydająca leki lub wyroby medyczne, oprócz obowiązków wynikających z art. 44, ma obowiązek poinformować świadczeniobiorcę o możliwości nabycia leku innego niż lek przepisany na recepcie, o tej samej nazwie międzynarodowej, dawce, postaci farmaceutycznej, która nie powoduje powstania różnic terapeutycznych, i o tym samym wskazaniu lub wyrobu medycznego mającego takie samo przewidziane zastosowanie oraz właściwości, objętego wykazem, o którym mowa w art. 37 ust. 1, w części dotyczącej bezpłatnego zaopatrzenia świadczeniobiorców w okresie ciąży albo połogu oraz ma obowiązek na żądanie świadczeniobiorcy wydać ten lek lub wyrób.”,
b) w ust. 3 wyrazy „Przepisy art. 44 ust. 1-2d” zastępuje się wyrazami „Przepisy art. 44 ust. 1-2c”;
50) użyte w art. 45 w ust. 2 w zdaniu drugim oraz w art. 45b w ust. 1 w części wspólnej i w ust. 3 wyrazy „ministra właściwego do spraw zdrowia” zastępuje się wyrazami „Funduszu”;
51) w art. 46 dodaje się ust. 9 w brzmieniu:
„9. Fundusz do dnia 31 marca każdego roku zamieszcza w Biuletynie Informacji Publicznej Funduszu zestawienie zbiorcze o zrealizowanej refundacji z budżetu państwa za rok poprzedni dla każdej apteki i każdego podmiotu realizującego zlecenia na zaopatrzenie w wyroby medyczne.”;
52) w art. 48:
a) ust. 7a i 7b otrzymują brzmienie:
„7a. Osoba uprawniona, z wyłączeniem lekarza ubezpieczenia zdrowotnego, felczera ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, pielęgniarki ubezpieczenia zdrowotnego i położnej ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, o których mowa w art. 15a ust. 1 i 2 ustawy z dnia 15 lipca 2011 r. o zawodach pielęgniarki i położnej, jest obowiązana do zwrotu Funduszowi kwoty stanowiącej równowartość kwoty refundacji wraz z odsetkami ustawowymi liczonymi od dnia dokonania refundacji, o której mowa w art. 46 ust. 1, w przypadku:
1) wypisania recepty w okresie pozbawienia prawa wystawiania recept, o którym mowa w ust. 5,
2) wypisania recepty nieuzasadnionej udokumentowanymi względami medycznymi,
3) wypisania recepty niezgodnej z uprawnieniami świadczeniobiorcy albo osoby uprawnionej,
4) wypisania recepty niezgodnej ze wskazaniami zawartymi w obwieszczeniach, o których mowa w art. 37 ust. 1 lub 4
- w terminie 14 dni od dnia otrzymania wezwania do zapłaty.
7b. Osoba uprawniona, z wyłączeniem lekarza ubezpieczenia zdrowotnego, felczera ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, pielęgniarki ubezpieczenia zdrowotnego i położnej ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, o których mowa w art. 15a ust. 1 i 2 ustawy z dnia 15 lipca 2011 r. o zawodach pielęgniarki i położnej, jest obowiązana do zwrotu Funduszowi kwoty stanowiącej równowartość limitu finansowania wyrobu medycznego wraz z odsetkami ustawowymi liczonymi od dnia dokonania finansowania, o którym mowa w przepisach wydanych na podstawie art. 38 ust. 4, w przypadku:
1) wypisania zlecenia w okresie pozbawienia prawa wystawiania recept, o którym mowa w ust. 5,
2) wypisania zlecenia nieuzasadnionego udokumentowanymi względami medycznymi,
3) wypisania zlecenia niezgodnego z kryteriami przyznawania zawartymi w przepisach, o których mowa w art. 38 ust. 4
- w terminie 14 dni od dnia otrzymania wezwania do zapłaty.”,
b) po ust. 7d dodaje się ust. 7e-7g w brzmieniu:
„7e. Osoba uprawniona, z wyłączeniem lekarza ubezpieczenia zdrowotnego, felczera ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, pielęgniarki ubezpieczenia zdrowotnego i położnej ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, o których mowa w art. 15a ust. 1 i 2 ustawy z dnia 15 lipca 2011 r. o zawodach pielęgniarki i położnej, która wystawiła receptę na refundowany lek, środek spożywczy specjalnego przeznaczenia żywieniowego albo wyrób medyczny, albo podmiot wykonujący działalność leczniczą, w ramach której wystawiono receptę na refundowany lek, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny albo zlecenie na wyrób medyczny, w przypadku:
1) uniemożliwienia czynności kontrolnych, o których mowa w dziale IIIA ustawy o świadczeniach,
2) niewykonania w terminie zaleceń pokontrolnych
- są obowiązani do zapłaty Funduszowi kary pieniężnej w wysokości równowartości kwoty refundacji za okres objęty czynnościami, o których mowa w dziale IIIA ustawy o świadczeniach, nieprzekraczający 5 lat liczonych od zakończenia roku kalendarzowego, w którym nastąpiła refundacja.
7f. Wysokość kary, o której mowa w ust. 7e, może zostać zmniejszona w sytuacji stwierdzenia w wyniku kontroli zasadności wystawienia recepty z niższym poziomem odpłatności. W takim przypadku karę pieniężną nakłada się w wysokości wynikającej z różnicy między wskazanym na recepcie poziomem odpłatności a poziomem, który w wyniku kontroli uznano za zasadny.
7g. W wezwaniu do zapłaty określa się każdorazowo wysokość kary, o której mowa w ust. 7e, oraz termin zapłaty, który nie może być krótszy niż 14 dni od dnia otrzymania wezwania.”;
53) uchyla się art. 52a;
54) w art. 53:
a) uchyla się ust. 2a,
b) w ust. 3 wyrazy „o których mowa w ust. 1, 2 oraz art. 52a” zastępuje się wyrazami „o których mowa w ust. 1 i 2”.
Art. 2. [Prawo farmaceutyczne]
1) w art. 2:
a) po pkt 72 dodaje się pkt 73 i 74 w brzmieniu:
„73) dyżurem w dzień wolny od pracy - jest dyżur pełniony przez aptekę ogólnodostępną w dniach, o których mowa w art. 1 lub art. 1a ust. 1 ustawy z dnia 18 stycznia 1951 r. o dniach wolnych od pracy (Dz. U. z 2020 r. poz. 1920), bez przerwy przez kolejne 4 godziny zegarowe w przedziale czasowym między godzinami 10.00 a 18.00;
74) dyżurem w porze nocnej - jest dyżur pełniony przez aptekę ogólnodostępną bez przerwy przez kolejne 2 godziny zegarowe w przedziale czasowym między godzinami 19.00 a 23.00;”,
b) pkt 7b otrzymuje brzmienie:
„7b) importem równoległym - jest każde działanie w rozumieniu art. 72 ust. 4 polegające na przywozie z państwa członkowskiego Unii Europejskiej lub państwa członkowskiego Europejskiego Porozumienia o Wolnym Handlu (EFTA) - strony umowy o Europejskim Obszarze Gospodarczym produktu leczniczego zawierającego tę samą substancję czynną lub to samo połączenie substancji czynnych, co najmniej te same wskazania do 3 poziomu kodu ATC/ATCvet (kod międzynarodowej klasyfikacji anatomiczno-terapeutyczno-chemicznej produktów leczniczych/kod międzynarodowej klasyfikacji anatomiczno-terapeutyczno-chemicznej produktów leczniczych weterynaryjnych), tę samą moc, tę samą drogę podania oraz tę samą postać jak produkt leczniczy dopuszczony do obrotu na terytorium Rzeczypospolitej Polskiej lub postać zbliżoną, która nie powoduje powstania różnic terapeutycznych w stosunku do produktu leczniczego dopuszczonego do obrotu na terytorium Rzeczypospolitej Polskiej;”,
c) pkt 35a otrzymuje brzmienie:
„35a) przedstawicielem podmiotu odpowiedzialnego - jest osoba fizyczna albo osoba prawna, mająca miejsce zamieszkania albo siedzibę na terytorium Rzeczypospolitej Polskiej, wyznaczona przez podmiot odpowiedzialny do stałego wykonywania jego obowiązków i uprawnień na terytorium Rzeczypospolitej Polskiej na podstawie umowy;”;
2) w art. 2a dodaje się ust. 5 w brzmieniu:
„5. Ilekroć w niniejszej ustawie jest mowa o:
1) zarządzie powiatu, rozumie się przez to również prezydenta miasta na prawach powiatu;
2) uchwale zarządu powiatu, rozumie się przez to również zarządzenie prezydenta miasta na prawach powiatu;
3) dyżurze bez bliższego określenia, rozumie się przez to dyżur w dzień wolny od pracy lub dyżur w porze nocnej.”;
3) w art. 4c ust. 1 otrzymuje brzmienie:
„1. W przypadkach uzasadnionych ochroną zdrowia publicznego Prezes Urzędu, uwzględniając bezpieczeństwo stosowania produktu leczniczego, może na czas określony wyrazić zgodę na zwolnienie:
1) z obowiązku umieszczania na opakowaniu lub w ulotce dołączanej do opakowania niektórych danych szczegółowych lub
2) w całości albo w części z obowiązku sporządzenia oznakowania opakowania lub ulotki dołączanej do opakowania w języku polskim
- dotyczącą produktu leczniczego, który posiada kategorię dostępności, o której mowa w art. 23a ust. 1 pkt 3 lub 5, lub innego produktu leczniczego, względem którego występują poważne trudności w zakresie jego dostępności.”;
4) w art. 21a ust. 3a otrzymuje brzmienie:
„3a. W przypadku wygaśnięcia pozwolenia na dopuszczenie do obrotu produktu leczniczego w państwie członkowskim Unii Europejskiej lub państwie członkowskim Europejskiego Porozumienia o Wolnym Handlu (EFTA) - stronie umowy o Europejskim Obszarze Gospodarczym, z którego produkt ten jest importowany równolegle, pozwolenie na import równoległy wygasa z dniem wygaśnięcia tego pozwolenia.”;
5) w art. 37azg ust. 2 otrzymuje brzmienie:
„2. W przypadku zagrożenia brakiem dostępności na terytorium Rzeczypospolitej Polskiej:
1) produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego lub produktu biobójczego, w związku ze stanem zagrożenia epidemicznego, stanem epidemii albo w razie niebezpieczeństwa szerzenia się zakażenia lub choroby zakaźnej, które może stanowić zagrożenie dla zdrowia publicznego, w szczególności wystąpienia choroby szczególnie niebezpiecznej i wysoce zakaźnej w rozumieniu art. 2 pkt 4 ustawy z dnia 5 grudnia 2008 r. o zapobieganiu oraz zwalczaniu zakażeń i chorób zakaźnych u ludzi, lub
2) produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego umieszczonego w:
a) obwieszczeniu, o którym mowa w art. 37av ust. 14, lub
b) obwieszczeniu, o którym mowa w art. 37 ust. 1 ustawy z dnia 12 maja 2011 r. o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych
- minister właściwy do spraw zdrowia może ograniczyć, w drodze obwieszczenia, ilość, jaką maksymalnie można wydać danemu pacjentowi w określonym czasie lub w określonym wskazaniu.”;
6) w art. 54 ust. 5 otrzymuje brzmienie:
„5. Reklama produktu leczniczego polegająca na bezpłatnym dostarczaniu jego próbek nie może dotyczyć produktów leczniczych zawierających środki odurzające lub substancje psychotropowe, lub prekursory kategorii 1, a w przypadku gdy odbiorcą takiej reklamy jest farmaceuta, nie może ona również dotyczyć produktów leczniczych o kategorii dostępności innej niż określona w art. 23a ust. 1 pkt 1.”;
7) w art. 81 po ust. 1 dodaje się ust. 1a w brzmieniu:
„1a. W przypadku, o którym mowa w ust. 1 pkt 2 lit. b, Główny Inspektor Farmaceutyczny może odstąpić od cofnięcia zezwolenia na prowadzenie hurtowni farmaceutycznej, jeżeli stwierdzi, że waga naruszenia prawa jest znikoma.”;
8) w art. 87 po ust. 2a dodaje się ust. 2b i 2c w brzmieniu:
„2b. Za zgodą wojewódzkiego inspektora farmaceutycznego właściwego ze względu na położenie zakładu leczniczego apteki szpitalnej nie tworzy się w zakładzie leczniczym, w którym podmiot leczniczy wykonuje działalność leczniczą w rodzaju całodobowe świadczenia zdrowotne inne niż szpitalne wyłącznie w zakresie świadczeń, o których mowa w art. 15 ust. 2 pkt 4-6 lub 11 ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych (Dz. U. z 2022 r. poz. 2561, z późn. zm.4)) - jeżeli udzielanie tych świadczeń nie wymaga stosowania produktów leczniczych wymagających nadzoru farmaceuty.
2c. Zgodę, o której mowa w ust. 2b, wydaje się na wniosek podmiotu leczniczego w drodze decyzji.”;
9) w art. 87a:
a) w ust. 1:
- w pkt 1 w lit. b skreśla się wyraz „rozpoczęte”,
- pkt 2 otrzymuje brzmienie:
„2) dziale farmacji szpitalnej:
a) równoważnik co najmniej 1 etatu w pełnym wymiarze godzin kierownika tego działu,
b) równoważnik co najmniej 1 etatu w pełnym wymiarze godzin co najmniej jednego farmaceuty w przypadku podmiotów posiadających co najmniej 100 łóżek lub stanowisk dializacyjnych zgłoszonych w rejestrze podmiotów wykonujących działalność leczniczą, o którym mowa w art. 106 ust. 1 ustawy z dnia 15 kwietnia 2011 r. o działalności leczniczej, zakładu leczniczego zaopatrywanego przez ten dział.”,
b) po ust. 1 dodaje się ust. 1a i 1b w brzmieniu:
„1a. Za zgodą wojewódzkiego inspektora farmaceutycznego właściwego ze względu na położenie zakładu leczniczego do liczby łóżek, o których mowa w ust. 1 pkt 1 lit. b lub pkt 2 lit. b, nie wlicza się łóżek służących udzielaniu świadczeń zdrowotnych, o których mowa w art. 15 ust. 2 pkt 4-6 lub 11 ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych - jeżeli udzielanie tych świadczeń nie wymaga stosowania produktów leczniczych wymagających nadzoru farmaceuty.
1b. Zgodę, o której mowa w ust. 1a, wydaje się na wniosek podmiotu leczniczego w drodze decyzji.”;
10) art. 94 otrzymuje brzmienie:
„Art. 94. 1. Rozkład godzin pracy aptek ogólnodostępnych znajdujących się na terenie powiatu jest dostosowany do potrzeb ludności tego powiatu i pozwala na realizację przez te apteki zadań określonych w art. 87 ust. 2, w szczególności przez pełnienie dyżurów w dzień wolny od pracy oraz dyżurów w porze nocnej, z wyłączeniem powiatu mającego swoją siedzibę w sąsiadującym mieście na prawach powiatu.
2. Podmiot prowadzący aptekę ogólnodostępną przekazuje zarządowi powiatu, w którym znajduje się ta apteka, rozkład godzin pracy tej apteki na dany rok najpóźniej do dnia 30 września roku poprzedzającego rok, w którym ma być podjęta uchwała, o której mowa w ust. 3, oraz informuje zarząd powiatu o zmianach tego rozkładu z co najmniej 30-dniowym wyprzedzeniem.
3. W przypadku gdy praca aptek ogólnodostępnych na terenie powiatu, którego siedzibą jest miasto liczące nie więcej niż 40 tys. mieszkańców, zgodna z przekazanymi na podstawie ust. 2 rozkładami godzin ich pracy, nie gwarantuje zaspokojenia potrzeb ludności, o których mowa w ust. 1, w porze nocnej oraz w dni wolne od pracy w zakresie zadań określonych w art. 87 ust. 2, zarząd powiatu wyznacza, w drodze uchwały, aptekę ogólnodostępną do pełnienia dyżurów w porze nocnej lub dyżurów w dni wolne od pracy. Uchwała jest wydawana na okres jednego roku kalendarzowego i określa dni oraz godziny pełnienia dyżurów przez aptekę ogólnodostępną prowadzoną przez określonego przedsiębiorcę.
4. Zarząd powiatu informuje podmioty prowadzące apteki ogólnodostępne na terenie tego powiatu, indywidualnie albo zbiorowo, w sposób określony w ust. 6, co najmniej na 21 dni przed dniem wydania uchwały, o której mowa w ust. 3, o potrzebie wyznaczenia apteki ogólnodostępnej do pełnienia dyżurów w porze nocnej lub dyżurów w dni wolne od pracy, wskazując dni i godziny, w jakich mają być pełnione te dyżury.
5. Podmiot prowadzący aptekę ogólnodostępną może zgłosić gotowość do pełnienia dyżurów przez prowadzoną przez siebie aptekę, we wskazanych przez siebie dniach i godzinach zgodnych z dniami i godzinami określonymi na podstawie ust. 4, w terminie 7 dni od dnia doręczenia informacji, o której mowa w ust. 4. Wraz ze zgłoszeniem osoba uprawniona do reprezentowania podmiotu prowadzącego aptekę ogólnodostępną składa w jego imieniu oświadczenie, że nie zachodzą okoliczności, o których mowa w ust. 8, oraz że w zgłaszanej aptece ogólnodostępnej spełnione są wymagania, o których mowa w ust. 7 pkt 3. Oświadczenie składa się pod rygorem odpowiedzialności karnej za składanie fałszywych oświadczeń przewidzianej w art. 233 § 6 ustawy z dnia 6 czerwca 1997 r. - Kodeks karny. Składający oświadczenie jest obowiązany do zawarcia w nim klauzuli następującej treści: „Jestem świadomy odpowiedzialności karnej za złożenie fałszywego oświadczenia.”. Klauzula ta zastępuje pouczenie organu o odpowiedzialności karnej za składanie fałszywych oświadczeń.
6. Komunikację między zarządem powiatu a podmiotami prowadzącymi apteki ogólnodostępne w sprawach, o których mowa w ust. 2, 4, 5 lub 12, prowadzi się za pośrednictwem środków komunikacji elektronicznej.
7. Zarząd powiatu dokonuje wyboru apteki ogólnodostępnej, która zostanie wyznaczona do pełnienia dyżurów w porze nocnej lub dyżurów w dni wolne od pracy, biorąc pod uwagę:
1) potrzeby mieszkańców powiatu w zakresie zaopatrzenia w produkty lecznicze, w tym leki recepturowe, wyroby medyczne oraz pozostałe produkty aptek ogólnodostępnych oraz w zakresie świadczenia usług farmaceutycznych i sprawowania opieki farmaceutycznej;
2) lokalne uwarunkowania geograficzne i demograficzne oraz położenie lokalu apteki ogólnodostępnej;
3) możliwość zapewnienia przez podmiot prowadzący aptekę ogólnodostępną asortymentu określonego w pkt 1 oraz liczby personelu dostosowanej do zapewniania potrzeb określonych w pkt 1.
8. Do pełnienia dyżurów w porze nocnej lub dyżurów w dni wolne od pracy nie wyznacza się apteki ogólnodostępnej, jeżeli wobec podmiotu, który ją prowadzi:
1) toczy się postępowanie administracyjne w przedmiocie cofnięcia zezwolenia na prowadzenie apteki;
2) w okresie 3 lat poprzedzających rok, w którym ma być pełniony dyżur, wydano:
a) ostateczną decyzję w przedmiocie cofnięcia zezwolenia na prowadzenie apteki ogólnodostępnej,
b) ostateczną decyzję w przedmiocie nakazania usunięcia w ustalonym terminie stwierdzonych uchybień w prowadzeniu działalności objętej zezwoleniem na prowadzenie apteki ogólnodostępnej,
c) prawomocne orzeczenie zakazujące wykonywania działalności gospodarczej objętej zezwoleniem na prowadzenie apteki ogólnodostępnej.
9. Zarząd powiatu:
1) wyznaczając aptekę ogólnodostępną do pełnienia dyżurów, dokonuje wyboru spośród aptek ogólnodostępnych, które zostały zgłoszone w sposób, o którym mowa w ust. 5;
2) w przypadku braku zgłoszenia apteki ogólnodostępnej w sposób, o którym mowa w ust. 5, lub w przypadku zgłoszenia apteki ogólnodostępnej niespełniającej kryteriów, o których mowa w ust. 7, wyznacza do pełnienia dyżurów inną aptekę ogólnodostępną, biorąc pod uwagę kryteria określone w ust. 7 i 8;
3) w przypadku gdy po przeprowadzonej przez siebie analizie poziomu zaspokajania potrzeb, o których mowa w ust. 1, zwłaszcza weryfikacji funkcjonowania na terenie powiatu przynajmniej jednej apteki ogólnodostępnej w trybie całodobowym, dokonanej w oparciu o informacje, o których mowa w ust. 2, uzna, że uzasadnione jest wyznaczenie:
a) apteki ogólnodostępnej do pełnienia dyżurów w mieście będącym siedzibą powiatu, liczącym więcej niż 40 tys. mieszkańców lub
b) szerszego niż określono w art. 2 pkt 73 lub 74 zakresu godzinowego dyżurów pełnionych przez wyznaczoną aptekę ogólnodostępną
- może dokonać takiego wyznaczenia.
10. Przed podjęciem uchwały, o której mowa w ust. 3, osoba uprawniona do reprezentowania podmiotu prowadzącego aptekę ogólnodostępną, o której mowa w ust. 9 pkt 2, w terminie 7 dni od dnia wezwania przez zarząd powiatu, składa oświadczenie o niezachodzeniu okoliczności, o których mowa w ust. 8, oraz że w wyznaczanej aptece ogólnodostępnej spełnione są wymagania, o których mowa w ust. 7 pkt 3. Oświadczenie składa się pod rygorem odpowiedzialności karnej za składanie fałszywych oświadczeń przewidzianej w art. 233 § 6 ustawy z dnia 6 czerwca 1997 r. - Kodeks karny. Składający oświadczenie jest obowiązany do zawarcia w nim klauzuli następującej treści: „Jestem świadomy odpowiedzialności karnej za złożenie fałszywego oświadczenia.”. Klauzula ta zastępuje pouczenie organu o odpowiedzialności karnej za składanie fałszywych oświadczeń.
11. W przypadku uzasadnionych wątpliwości w zakresie prawdziwości okoliczności wskazanych w oświadczeniach, o których mowa w ust. 5 lub 10, zarząd powiatu może wystąpić do wojewódzkiego inspektora farmaceutycznego z wnioskiem o potwierdzenie tych okoliczności. Wojewódzki inspektor farmaceutyczny udziela zarządowi powiatu odpowiedzi niezwłocznie, jednak nie później niż w terminie 14 dni od dnia otrzymania wniosku.
12. Zarząd powiatu niezwłocznie przekazuje kopię uchwały, o której mowa w ust. 3, lub jej zmian, w przypadku, o którym mowa w ust. 21, właściwemu miejscowo wojewódzkiemu inspektorowi farmaceutycznemu, dyrektorowi oddziału wojewódzkiego Narodowego Funduszu Zdrowia oraz aptece ogólnodostępnej wyznaczonej do pełnienia dyżurów.
13. Pełnienie dyżurów na podstawie uchwały, o której mowa w ust. 3, przez aptekę ogólnodostępną podlega finansowaniu ze środków Narodowego Funduszu Zdrowia, z wyjątkiem pełnienia ich w zakresie godzinowym rozszerzonym zgodnie z ust. 9 pkt 3 lit. b - w takim przypadku powiat finansuje, z własnych środków, pełnienie tych dyżurów w zakresie wykraczającym ponad zakres godzinowy wynikający z art. 2 pkt 73 lub 74.
14. Finansowanie dyżurów aptek ogólnodostępnych, o których mowa w ust. 13, ma postać wynagrodzenia ryczałtowego w kwocie stanowiącej równowartość 3,5% minimalnego wynagrodzenia za pracę określonego na podstawie przepisów o minimalnym wynagrodzeniu za pracę, w przeliczeniu na 1 godzinę faktycznie przeprowadzonego dyżuru.
15. W celu realizacji uchwały, o której mowa w ust. 3, podmiot prowadzący aptekę ogólnodostępną zawiera umowę z Narodowym Funduszem Zdrowia na okres obowiązywania tej uchwały.
16. Prezes Narodowego Funduszu Zdrowia zamieszcza wzór umowy, o której mowa w ust. 15, w Biuletynie Informacji Publicznej Narodowego Funduszu Zdrowia. Apteka ogólnodostępna przekazuje do właściwego oddziału wojewódzkiego Narodowego Funduszu Zdrowia zestawienie godzin dyżurów odbytych w danym okresie rozliczeniowym, opracowywane zgodnie z umową, o której mowa w ust. 15, wraz z zestawieniem zbiorczym, o którym mowa w art. 45 ust. 5 ustawy z dnia 12 maja 2011 r. o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych.
17. Właściwy oddział wojewódzki Narodowego Funduszu Zdrowia wypłaca wynagrodzenie, o którym mowa w ust. 14, na rachunek bankowy podmiotu prowadzącego aptekę ogólnodostępną nie później niż w terminie 15 dni od dnia otrzymania zestawienia godzin dyżurów, o którym mowa w ust. 16 zdanie drugie.
18. Uchwała, o której mowa w ust. 3, nie stanowi aktu prawa miejscowego.
19. Ustalanie liczby mieszkańców miasta:
1) stanowiącego siedzibę powiatu,
2) na prawach powiatu
- następuje według stanu wskazanego w dokumencie pt. „Powierzchnia i ludność w przekroju terytorialnym” opracowywanym przez Główny Urząd Statystyczny i publikowanym na stronie internetowej Głównego Urzędu Statystycznego, aktualnym na dzień 30 września roku poprzedzającego rok kalendarzowy, na który ma zostać podjęta uchwała, o której mowa w ust. 3.
20. Jeżeli w wyniku kontroli, o której mowa w art. 47 ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych, zostanie stwierdzone, że apteka ogólnodostępna nie pełniła dyżuru albo pełniła go w wymiarze mniejszym, niż zostało wyznaczone, podmiot prowadzący aptekę ma obowiązek zwrotu dotychczas otrzymanego wynagrodzenia ryczałtowego z tytułu pełnienia dyżurów, od czasu wyznaczenia do ich pełnienia, wraz z odsetkami ustawowymi za opóźnienie, w terminie 14 dni od dnia otrzymania wezwania do zapłaty.
21. W przypadku gdy z przyczyn losowych wyznaczona do pełnienia dyżurów apteka ogólnodostępna traci możliwość ich pełnienia, zawiadamia o tym zarząd powiatu, który wyznacza inną aptekę ogólnodostępną do pełnienia dyżurów. W takim przypadku:
1) umowa, o której mowa w ust. 15, zawarta z pierwotnie wyznaczoną apteką ogólnodostępną wygasa z dniem skutecznego poinformowania Narodowego Funduszu Zdrowia przez zarząd powiatu o podjęciu uchwały, o której mowa w pkt 2, przy czym finansowanie tej aptece dyżurów następuje za okres ich faktycznego pełnienia;
2) zarząd powiatu wydaje uchwałę zmieniającą uchwałę, o której mowa w ust. 3, w zakresie wskazania w niej nowo wyznaczanej do pełnienia dyżurów apteki ogólnodostępnej, przy czym okres obowiązywania zmienianej uchwały pozostaje bez zmian;
3) wyznaczenia nowej apteki ogólnodostępnej do pełnienia dyżurów dokonuje się na podstawie informacji uzyskanych w procedurze, o której mowa w ust. 4 i 5, przeprowadzonej przed wyznaczeniem apteki ogólnodostępnej, która zaprzestała pełnienia dyżurów, przy czym w pierwszej kolejności, jeżeli to możliwe, wyboru dokonuje się spośród aptek ogólnodostępnych dobrowolnie zgłoszonych na podstawie ust. 5.”;
11) w art. 95b po ust. 1 dodaje się ust. 1a w brzmieniu:
„1a. Recepta, na której ma zostać przepisany produkt leczniczy o:
1) kategorii dostępności, o której mowa w art. 23a ust. 1 pkt 4,
2) innej kategorii dostępności niż określona w pkt 1, zawierający w swoim składzie substancję psychotropową lub środek odurzający, z których którakolwiek substancja lub środek należy do grupy II-N, III-P albo IV-P określonej w ustawie z dnia 29 lipca 2005 r. o przeciwdziałaniu narkomanii
- jest wystawiana wyłącznie w postaci elektronicznej.”;
12) w art. 96a:
a) w ust. 1d pkt 2 otrzymuje brzmienie:
„2) informację o zasadności wydania pacjentowi określonego produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego przez zapis „nie zamieniać” lub „NZ” przy pozycji produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, którego dotyczy ta informacja, pod warunkiem uzasadnionych wskazań medycznych lub występujących przy zastosowaniu tego produktu leczniczego udokumentowanych działań niepożądanych.”,
b) po ust. 1d dodaje się ust. 1da i 1db w brzmieniu:
„1da. W przypadku uwzględnienia na recepcie informacji, o której mowa w ust. 1d pkt 2, osoba wystawiająca receptę dodatkowo zamieszcza w dokumentacji medycznej pacjenta, którego recepta dotyczy, szczegółowe uzasadnienie przepisania temu pacjentowi określonego produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego.
1db. Niezależnie od ust. 1d pkt 2, na żądanie lub za zgodą pacjenta dopuszcza się wydanie:
1) innego produktu leczniczego niż przepisany przez osobę wystawiającą receptę - jeżeli całkowita ilość substancji czynnej albo substancji czynnych, zawartych w wydawanym produkcie leczniczym odpowiada całkowitej ilości substancji czynnej albo substancji czynnych zawartych w przepisanym na recepcie produkcie leczniczym, a produkty te posiadają również te same wskazania i tę samą drogę podania;
2) innego niż przepisany na recepcie, środka spożywczego specjalnego przeznaczenia żywieniowego mającego ten sam albo zbliżony skład, zastosowanie lub sposób przygotowania, lub wyrobu medycznego mającego takie samo przewidziane zastosowanie oraz właściwości.”,
c) w ust. 7a pkt 2 otrzymuje brzmienie:
„2) pacjent od dnia realizacji recepty może otrzymać maksymalnie ilość produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego niezbędną pacjentowi do 120-dniowego okresu stosowania wyliczonego na podstawie określonego na recepcie sposobu dawkowania”,
d) po ust. 7a dodaje się ust. 7aa i 7ab w brzmieniu:
„7aa. W przypadku gdy na recepcie, o której mowa w ust. 7a pkt 2, przepisano ilość większą niż do 120-dniowego stosowania, kolejną ilość produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego niezbędną do następnego 120-dniowego okresu stosowania pacjent może otrzymać po upływie 3/4 okresu, na który zrealizował receptę.
7ab. Obliczenia ilości produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego przeznaczonego do wydania pacjentowi na zasadach, o których mowa w ust. 7a i 7aa, z wyjątkiem ilości leku recepturowego oraz ilości produktu leczniczego, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego przeznaczonego do stosowania na okres nie dłuższy niż 120 dni, dokonuje system teleinformatyczny, o którym mowa w art. 7 ust. 1 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia.”;
13) w art. 103:
a) po ust. 1a dodaje się ust. 1b w brzmieniu:
„1b. W przypadku, o którym mowa w ust. 1 pkt 2, wojewódzki inspektor farmaceutyczny może odstąpić od cofnięcia zezwolenia na prowadzenie apteki ogólnodostępnej, jeżeli stwierdzi, że waga naruszenia prawa jest znikoma.”,
b) w ust. 2 w pkt 10 kropkę zastępuje się średnikiem i dodaje pkt 11 w brzmieniu:
„11) apteka zbywa produkty lecznicze, środki spożywcze specjalnego przeznaczenia żywieniowego lub wyroby medyczne niezgodnie z art. 37azg ust. 2.”;
14) po art. 104a dodaje się art. 104b w brzmieniu:
„Art. 104b. W przypadku cofnięcia zezwolenia na prowadzenie apteki lub punktu aptecznego, jego przeniesienia, uchylenia, stwierdzenia wygaśnięcia albo nieważności wojewódzki inspektor farmaceutyczny, w terminie 3 dni od dnia, w którym uzyska informację, że decyzja o cofnięciu zezwolenia na prowadzenie apteki lub punktu aptecznego, jego przeniesieniu, uchyleniu, stwierdzeniu wygaśnięcia albo nieważności stała się ostateczna, przekazuje tę informację w postaci elektronicznej oddziałowi wojewódzkiemu Narodowego Funduszu Zdrowia.”;
15) w art. 109 w pkt 15 kropkę zastępuje się średnikiem i dodaje się pkt 16 w brzmieniu:
„16) współpraca z zarządami powiatów w sprawach, o których mowa w art. 94 ust. 11.”;
16) po art. 129ba dodaje się art. 129bb w brzmieniu:
„Art. 129bb. 1. Karze pieniężnej w wysokości do 2000 zł podlega podmiot prowadzący aptekę ogólnodostępną:
1) który nie przekazał informacji, o której mowa w art. 94 ust. 2, lub
2) w imieniu którego nie zostało złożone oświadczenie, o którym mowa w art. 94 ust. 10 zdanie pierwsze.
2. Karę pieniężną, o której mowa w ust. 1, nakłada w drodze decyzji starosta. Przepisy art. 129ba ust. 1 i 2 stosuje się.
3. Starosta jest wierzycielem w rozumieniu przepisów o postępowaniu egzekucyjnym w administracji.
4. Wpływy z kar pieniężnych, o których mowa w ust. 1, wraz z odsetkami za zwłokę w wysokości jak dla zaległości podatkowych stanowią dochód powiatu.”.
Art. 3. [Ustawa o pomocy społecznej]
Art. 4. [Ustawa o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych]
1) w art. 31lb:
a) ust. 5 otrzymuje brzmienie:
„5. Osoby, które zgłaszają uwagi do opublikowanego raportu w sprawie ustalenia taryfy świadczeń, składają oświadczenie, o którym mowa w art. 31sa ust. 8a. Oświadczenie składa się wraz ze zgłoszonymi uwagami do raportu w sprawie ustalenia taryfy świadczeń.”,
b) w ust. 7:
- we wprowadzeniu do wyliczenia w zdaniu pierwszym wyrazy „o których mowa w ust. 4” zastępuje się wyrazami „o których mowa w ust. 4 zdanie drugie”,
- część wspólna otrzymuje brzmienie:
„- jeżeli te osoby lub podmioty złożyły przed posiedzeniem Rady do spraw Taryfikacji oświadczenia, o których mowa w art. 31sa ust. 8a.”,
c) po ust. 7 dodaje się ust. 7a-7c w brzmieniu:
„7a. Przewodniczący lub wiceprzewodniczący Rady do spraw Taryfikacji analizuje złożone oświadczenia, o których mowa w ust. 7, kierując się zasadą równego traktowania osób składających oświadczenia oraz biorąc pod uwagę rodzaj zgłoszonego powiązania branżowego, jego charakter i okoliczności zgłoszonego powiązania branżowego, a także ewentualny wpływ zadeklarowanego powiązania branżowego na postępowanie prowadzone przez Radę do spraw Taryfikacji. Analiza jest wykonywana w oparciu o sposób i tryb określone w przepisach wydanych na podstawie ust. 7c, w celu zapewnienia obiektywności rozstrzygnięć podejmowanych przez Radę i efektywności prowadzonych przez nią postępowań oraz niedopuszczenia do udziału w konsultacjach osób lub wyłączenia z konsultacji osób, w stosunku do których ujawnił się konflikt interesów budzący uzasadnione wątpliwości co do ich bezstronności.
7b. Przewodniczący lub wiceprzewodniczący Rady do spraw Taryfikacji na podstawie dokonanej analizy, o której mowa w ust. 7a, dopuszcza do udziału w konsultacjach, odmawia dopuszczenia do udziału w konsultacjach lub wyłącza z konsultacji osoby, o których mowa w ust. 7.
7c. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, sposób i tryb analizy składanych oświadczeń, o których mowa w ust. 7 i art. 31sa ust. 10 i 11, oraz składanych deklaracji, o których mowa w art. 31s ust. 12 i 15, uwzględniając konieczność oceny bezstronności osób biorących udział w prowadzonych postępowaniach.”;
2) w art. 31n po pkt 2c dodaje się pkt 2d-2i w brzmieniu:
„2d) przygotowywanie opinii dotyczących opisów programów lekowych, o których mowa w art. 16a ust. 1 ustawy o refundacji;
2e) przekazywanie ministrowi właściwemu do spraw zdrowia wykazu, o którym mowa w art. 30a ust. 2 ustawy o refundacji;
2f) przygotowywanie oceny dokumentacji, o której mowa w art. 35 ust. 8c ustawy o refundacji; 2g) przygotowywanie opinii, o których mowa w art. 15 ust. 3 ustawy o refundacji;
2h) przygotowywanie raportów z oceny efektywności objętych refundacją technologii lekowych o wysokim poziomie innowacyjności oraz jakości leczenia, o których mowa w art. 40a ust. 7 ustawy o refundacji;
2i) przygotowywanie co 5 lat raportu w sprawie oceny technologii medycznych stosowanych w procedurze zapłodnienia pozaustrojowego, o którym mowa w art. 71 pkt 4 ustawy z dnia 25 czerwca 2015 r. o leczeniu niepłodności (Dz. U. z 2020 r. poz. 442);”;
3) w art. 31o w ust. 2:
a) po pkt 1f dodaje się pkt 1g-1j w brzmieniu:
„1g) wydawanie opinii, o których mowa w art. 15 ust. 3 ustawy o refundacji;
1h) wydawanie opinii dotyczących opisów programów lekowych, o których mowa w art. 16a ust. 1 ustawy o refundacji;
1i) przygotowanie wykazu, o którym mowa w art. 30a ust. 2 ustawy o refundacji;
1j) ocena dokumentacji, o której mowa w art. 35 ust. 8c ustawy o refundacji;”,
b) w pkt 5:
- lit. f otrzymuje brzmienie:
„f) protokołów z posiedzeń Rady Przejrzystości i Rady do spraw Taryfikacji,”,
- w lit. j kropkę zastępuje się przecinkiem i dodaje się lit. k oraz l w brzmieniu:
„k) zaktualizowanej wysokości opłaty, o której mowa w art. 35 ust. 3 ustawy o refundacji,
l) wykazów na podstawie art. 30a ust. 2 ustawy o refundacji.”;
4) w art. 31q ust. 3 otrzymuje brzmienie:
„3. Nabór kandydatów do zatrudnienia w Agencji jest otwarty i konkurencyjny. Przepisy art. 107a ust. 2 i art. 107b-107g stosuje się odpowiednio.”;
5) w art. 31s:
a) ust. 4 otrzymuje brzmienie:
„4. Kadencja Rady Przejrzystości trwa 6 lat. W przypadku gdy członek Rady Przejrzystości zostanie odwołany przed upływem kadencji, kadencja członka powołanego na jego miejsce upływa z dniem upływu kadencji Rady Przejrzystości.”,
b) w ust. 6 pkt 5 otrzymuje brzmienie:
„5) wydawanie opinii w zakresie, o którym mowa w art. 30a ust. 5, art. 33 ust. 2, art. 40 oraz art. 40a ust. 5 i 8 ustawy o refundacji.”,
c) ust. 7 otrzymuje brzmienie:
„7. Członkiem Rady Przejrzystości może być wyłącznie osoba, której kandydatura została zaakceptowana przez ministra właściwego do spraw zdrowia, wobec której nie zachodzą okoliczności określone w ust. 8, oraz która:
1) posiada wiedzę i doświadczenie w zakresie świadczeń opieki zdrowotnej, dające rękojmię prawidłowego wykonywania obowiązków;
2) nie została skazana prawomocnym wyrokiem za umyślne przestępstwo lub przestępstwo skarbowe;
3) korzysta z pełni praw publicznych;
4) nie jest członkiem Rady do spraw Taryfikacji, o której mowa w art. 31sa, lub nie jest członkiem Komisji Ekonomicznej, o której mowa w art. 17 ustawy o refundacji;
5) nie pełni funkcji Prezesa Agencji albo jego zastępcy.”,
d) w ust. 8:
- pkt 2 otrzymuje brzmienie:
„2) być członkami organów spółek handlowych lub przedstawicielami przedsiębiorców prowadzących działalność gospodarczą w zakresie świadczenia usług prawnych, marketingowych lub doradczych związanych z wytwarzaniem, obrotem lub refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego;”,
- w pkt 5 kropkę zastępuje się średnikiem i dodaje się pkt 6 w brzmieniu:
„6) wykonywać zajęć zarobkowych na podstawie stosunku pracy, umowy o świadczenie usług zarządczych, umowy zlecenia, umowy o dzieło lub innej umowy o podobnym charakterze zawartej z podmiotami, o których mowa w pkt 1-3, chyba że złożą oświadczenie o wykonywaniu zajęć zarobkowych na rzecz określonych podmiotów i wskażą ich zakres.”,
e) po ust. 8 dodaje się ust. 8a-8c w brzmieniu:
„8a. Kandydaci na członków Rady Przejrzystości przed powołaniem do składu tej Rady składają ministrowi właściwemu do spraw zdrowia, a członkowie Rady Przejrzystości przed każdym posiedzeniem tej Rady składają Prezesowi Agencji, pod rygorem odpowiedzialności karnej za składanie fałszywych oświadczeń z art. 233 § 6 ustawy z dnia 6 czerwca 1997 r. - Kodeks karny, oświadczenie o:
1) zaistnieniu lub niezaistnieniu okoliczności określonych w ust. 8,
2) wykonywaniu lub niewykonywaniu zajęć zarobkowych na podstawie stosunku pracy, umowy o świadczenie usług zarządczych, umowy zlecenia, umowy o dzieło lub innej umowy o podobnym charakterze zawartej z podmiotami, o których mowa w ust. 8 pkt 1-3
- zwane dalej „deklaracją o powiązaniach branżowych”, dotyczące ich samych, ich małżonków, zstępnych i wstępnych w linii prostej oraz osób, z którymi pozostają we wspólnym pożyciu.
8b. Deklaracja o powiązaniach branżowych jest składana w przypadku:
1) kandydatów na członków Rady Przejrzystości za okres 3 lat poprzedzających dzień złożenia deklaracji;
2) członków Rady Przejrzystości za okres od dnia złożenia ostatniej deklaracji, w tym jako kandydatów na członków Rady, do dnia poprzedzającego posiedzenie Rady, w którym biorą udział;
3) osób, o których mowa w ust. 12, za okres jednego roku poprzedzającego dzień przyjęcia zlecenia;
4) osób, o których mowa w ust. 15, za okres jednego roku poprzedzającego dzień posiedzenia Rady;
5) osób, o których mowa w ust. 23, za okres jednego roku poprzedzającego dzień zgłoszenia uwag.
8c. Deklaracja o powiązaniach branżowych zawiera:
1) powód jej złożenia i wskazanie okresu, za jaki jest składana;
2) imię (imiona) i nazwisko osoby składającej deklarację;
3) numer PESEL osoby składającej deklarację, a jeżeli nie posiada - datę i miejsce urodzenia oraz obywatelstwo;
4) imię (imiona) i nazwisko: małżonka, wstępnych i zstępnych w linii prostej oraz osób, z którymi składający deklarację pozostaje we wspólnym pożyciu;
5) numery PESEL osób pozostających we wspólnym pożyciu z osobą składającą deklarację, a jeżeli nie posiadają - datę i miejsce urodzenia oraz obywatelstwo;
6) oświadczenie o następującej treści: „Jestem świadomy odpowiedzialności karnej za złożenie fałszywego oświadczenia.”. Klauzula ta zastępuje pouczenie organu o odpowiedzialności karnej za składanie fałszywych oświadczeń.”,
f) ust. 9 i 9a otrzymują brzmienie:
„9. Zaistnienie okoliczności, o których mowa w ust. 8 pkt 1-6, skutkuje odwołaniem członka ze składu Rady Przejrzystości.
9a. W przypadku niezłożenia przed posiedzeniem Rady Przejrzystości deklaracji o powiązaniach branżowych minister właściwy do spraw zdrowia odwołuje członka Rady Przejrzystości. Jeżeli złożenie deklaracji było niemożliwe z przyczyn technicznych lub wynikało z nieplanowanej potrzeby włączenia kolejnego członka Rady Przejrzystości do już trwającego posiedzenia, a członek Rady Przejrzystości zgłosi do protokołu zaistnienie okoliczności określonych w ust. 8 w celu ich oceny zgodnie z ust. 13, to deklarację uznaje się za skutecznie złożoną, pod warunkiem że wpłynie do przewodniczącego Rady Przejrzystości do końca dnia, w którym rozpoczęło się dane posiedzenie.”,
g) po ust. 9a dodaje się ust. 9b w brzmieniu:
„9b. W przypadku poświadczenia w deklaracji o powiązaniach branżowych nieprawdy minister właściwy do spraw zdrowia odwołuje członka Rady Przejrzystości.”,
h) ust. 12 otrzymuje brzmienie:
„12. Osoby niebędące członkami Rady Przejrzystości, którym zleca się przygotowanie ekspertyzy pisemnej lub ustnej lub innego opracowania dla Rady - albo w związku z przygotowywaniem ekspertyzy, rekomendacji lub opinii dla Prezesa Agencji, albo w związku z przygotowywaniem stanowiska lub opinii dla Rady - składają każdorazowo przed przyjęciem zlecenia deklarację o powiązaniach branżowych dla każdej przygotowanej ekspertyzy, rekomendacji, stanowiska lub opinii.”,
i) po ust. 12 dodaje się ust. 12a i 12b w brzmieniu:
„12a. Prezes Agencji analizuje deklaracje o powiązaniach branżowych złożone przez osoby, o których mowa w ust. 12, kierując się zasadą równego traktowania osób składających deklaracje oraz biorąc pod uwagę rodzaj zgłoszonego powiązania branżowego, jego charakter i okoliczności zgłoszonego powiązania branżowego, a także ewentualny wpływ zadeklarowanego powiązania branżowego na postępowanie prowadzone przez Radę Przejrzystości. Analiza jest wykonywana w oparciu o sposób i tryb określone w przepisach wydanych na podstawie art. 31lb ust. 7c, w celu zapewnienia obiektywności rozstrzygnięć podejmowanych przez Agencję, efektywności prowadzonych przez nią postępowań oraz niedopuszczania do udziału w tych postępowaniach osób, w stosunku do których ujawnił się konflikt interesów budzący uzasadnione wątpliwości co do ich bezstronności.
12b. Prezes Agencji na podstawie dokonanej analizy, o której mowa w ust. 12a, uwzględnia ekspertyzy pisemne lub ustne lub inne opracowanie dla Rady Przejrzystości lub Agencji albo ich nie uwzględnia.”,
j) ust. 13 otrzymuje brzmienie:
„13. W przypadku ujawnienia konfliktu interesów członka Rady Przejrzystości przewodniczący lub wiceprzewodniczący Rady wyłącza tego członka z udziału w pracach tej Rady oraz informuje o tym Prezesa Agencji.”,
k) po ust. 13 dodaje się ust. 13a w brzmieniu:
„13a. W przypadku gdy okoliczności, o których mowa w ust. 8a pkt 2, trwale uniemożliwiają członkowi Rady Przejrzystości właściwe wykonywanie przez niego zadań, minister właściwy do spraw zdrowia odwołuje tego członka Rady.”,
l) ust. 14 i 15 otrzymują brzmienie:
„14. W przypadku zaistnienia okoliczności, o których mowa w ust. 8 pkt 4, niezależnych od członka Rady Przejrzystości, informuje on niezwłocznie Prezesa Agencji o tym fakcie. Prezes Agencji zawiesza członka w pracach Rady Przejrzystości i wyznacza mu termin usunięcia zaistniałych okoliczności, a także informuje o tym fakcie ministra właściwego do spraw zdrowia. Po bezskutecznym upływie wyznaczonego terminu minister właściwy do spraw zdrowia odwołuje członka Rady Przejrzystości.
15. W posiedzeniach Rady Przejrzystości mogą brać udział eksperci z dziedziny medycyny, której dotyczą omawiane na danym posiedzeniu wnioski lub informacje, oraz inne osoby zaproszone przez przewodniczącego Rady Przejrzystości, bez prawa głosu. Osoby te, przed udziałem w posiedzeniu Rady, składają do Prezesa Agencji deklarację o powiązaniach branżowych.”,
m) po ust. 15 dodaje się ust. 15a i 15b w brzmieniu:
„15a. Przewodniczący lub wiceprzewodniczący Rady Przejrzystości analizuje deklaracje o powiązaniach branżowych złożone przez osoby, o których mowa w ust. 15, kierując się zasadą równego traktowania osób składających deklaracje oraz biorąc pod uwagę rodzaj zgłoszonego powiązania branżowego, jego charakter i okoliczności zgłoszonego powiązania branżowego, a także ewentualny wpływ zadeklarowanego powiązania branżowego na postępowanie prowadzone przez Radę Przejrzystości. Analiza jest wykonywana w oparciu o sposób i tryb określone w przepisach wydanych na podstawie art. 31lb ust. 7c, w celu zapewnienia obiektywności rozstrzygnięć podejmowanych przez Radę, efektywności prowadzonych przez nią postępowań oraz niedopuszczania do udziału w tych postępowaniach osób, w stosunku do których ujawnił się konflikt interesów budzący wątpliwości co do ich bezstronności.
15b. Przewodniczący lub wiceprzewodniczący Rady Przejrzystości na podstawie dokonanej analizy, o której mowa w ust. 15a, dopuszcza albo nie dopuszcza do udziału w posiedzeniu Rady ekspertów oraz inne osoby przez niego zaproszone.”,
n) ust. 21 otrzymuje brzmienie:
„21. Prezes Agencji albo Zespół bezwzględną większością głosów może zdecydować o konieczności podjęcia uchwały przez Radę Przejrzystości w pełnym składzie. Przepisy ust. 19 i 20 stosuje się odpowiednio.”,
o) ust. 23 i 24 otrzymują brzmienie:
„23. Deklarację o powiązaniach branżowych składają do Prezesa Agencji także osoby, które zgłaszają uwagi do upublicznionej analizy weryfikacyjnej Agencji. Deklaracja o powiązaniach branżowych jest składana wraz ze zgłaszanymi uwagami. Zgłoszone uwagi są rozpatrywane przez Agencję i zamieszczane w Biuletynie Informacji Publicznej Agencji wraz z wypełnioną deklaracją o powiązaniach branżowych.
24. Deklaracje o powiązaniach branżowych składane przez kandydatów na członków oraz członków Rady Przejrzystości weryfikuje Centralne Biuro Antykorupcyjne.”,
p) po ust. 24 dodaje się ust. 24a w brzmieniu:
„24a. Deklaracje o powiązaniach branżowych składane przez osoby inne niż określone w ust. 24 weryfikuje Centralne Biuro Antykorupcyjne z własnej inicjatywy lub na pisemny, uzasadniony wniosek Prezesa Agencji.”,
q) dodaje się ust. 26 w brzmieniu:
„26. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, wzór deklaracji o powiązaniach branżowych, mając na uwadze zapewnienie przejrzystości i komunikatywności tych deklaracji oraz potrzebę ujednolicenia ich formy.”;
6) w art. 31sa:
a) ust. 7 i 8 otrzymują brzmienie:
„7. Członkiem Rady do spraw Taryfikacji może być wyłącznie osoba, której kandydatura została zaakceptowana przez ministra właściwego do spraw zdrowia, wobec której nie zachodzą okoliczności określone w ust. 8, oraz która:
1) posiada wiedzę i doświadczenie w zakresie finansowania świadczeń opieki zdrowotnej, dające rękojmię prawidłowego wykonywania obowiązków;
2) nie została skazana prawomocnym wyrokiem za umyślne przestępstwo lub przestępstwo skarbowe;
3) korzysta z pełni praw publicznych;
4) nie jest członkiem Rady Przejrzystości lub nie jest członkiem Komisji Ekonomicznej, o której mowa w art. 17 ustawy o refundacji;
5) nie pełni funkcji Prezesa Agencji albo jego zastępcy.
8. Członkowie Rady do spraw Taryfikacji, ich małżonkowie, zstępni i wstępni w linii prostej oraz osoby, z którymi członkowie Rady pozostają we wspólnym pożyciu, nie mogą:
1) być członkami organów spółek handlowych lub przedstawicielami przedsiębiorców prowadzących działalność gospodarczą w zakresie doradztwa związanego z wyceną lub rozliczaniem świadczeń opieki zdrowotnej lub restrukturyzacji, lub audytu świadczeniodawców, lub ubezpieczeń zdrowia i życia;
2) być członkami organów spółek handlowych lub przedstawicielami przedsiębiorców prowadzących działalność gospodarczą w zakresie świadczenia usług prawnych, marketingowych lub doradczych dotyczących wytwarzania, obrotu lub refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego lub realizacji świadczeń opieki zdrowotnej, lub ubezpieczeń zdrowia i życia;
3) być członkami organów spółdzielni, stowarzyszeń lub fundacji prowadzących działalność, o której mowa w pkt 1 lub 2;
4) posiadać akcji lub udziałów w spółkach handlowych prowadzących działalność, o której mowa w pkt 1 lub 2, oraz udziałów w spółdzielniach prowadzących działalność, o której mowa w pkt 1 lub 2;
5) prowadzić działalności gospodarczej w zakresie, o którym mowa w pkt 1 lub 2;
6) wykonywać zajęć zarobkowych na podstawie stosunku pracy, umowy o świadczenie usług zarządczych, umowy zlecenia, umowy o dzieło lub innej umowy o podobnym charakterze zawartej z podmiotami, o których mowa w pkt 1 lub 2.”,
b) po ust. 8 dodaje się ust. 8a-8c w brzmieniu:
„8a. Kandydaci na członków Rady do spraw Taryfikacji przed powołaniem do składu tej Rady składają ministrowi właściwemu do spraw zdrowia, a członkowie Rady do spraw Taryfikacji przed każdym posiedzeniem tej Rady składają Prezesowi Agencji, pod rygorem odpowiedzialności karnej za składanie fałszywych oświadczeń z art. 233 § 6 ustawy z dnia 6 czerwca 1997 r. - Kodeks karny, oświadczenie o zaistnieniu lub niezaistnieniu okoliczności określonych w ust. 8, zwane dalej „oświadczeniem o powiązaniach branżowych”, dotyczące ich samych, ich małżonków, zstępnych i wstępnych w linii prostej oraz osób, z którymi pozostają we wspólnym pożyciu.
8b. Oświadczenie o powiązaniach branżowych jest składane w przypadku:
1) kandydatów na członków Rady do spraw Taryfikacji za okres 3 lat poprzedzających dzień złożenia oświadczenia;
2) członków Rady do spraw Taryfikacji za okres od dnia złożenia ostatniego oświadczenia, w tym jako kandydatów na członków Rady, do dnia poprzedzającego posiedzenie Rady, w którym biorą udział;
3) osób, o których mowa w ust. 10, za okres jednego roku poprzedzającego dzień przyjęcia zlecenia;
4) osób, o których mowa w ust. 11, za okres jednego roku poprzedzającego dzień posiedzenia Rady lub za okres jednego roku od dnia poprzedzającego przyjęcie zlecenia, o którym mowa w ust. 10;
5) osób, o których mowa w art. 31lb ust. 5, za okres jednego roku poprzedzającego dzień zgłoszenia uwag;
6) osób, o których mowa w art. 31lb ust. 7, za okres jednego roku poprzedzającego dzień posiedzenia Rady dotyczącego konsultacji w sprawie taryf świadczeń.
8c. Oświadczenie o powiązaniach branżowych zawiera:
1) powód jego złożenia i wskazanie okresu, za jaki jest składane;
2) imię (imiona) i nazwisko osoby składającej oświadczenie;
3) numer PESEL osoby składającej oświadczenie, a jeżeli nie posiada - datę i miejsce urodzenia oraz obywatelstwo;
4) imię (imiona) i nazwisko: małżonka, wstępnych i zstępnych w linii prostej oraz osób, z którymi składający oświadczenie pozostaje we wspólnym pożyciu;
5) numery PESEL osób pozostających we wspólnym pożyciu z osobą składającą oświadczenie, a jeżeli nie posiadają - datę i miejsce urodzenia oraz obywatelstwo;
6) oświadczenie o następującej treści: „Jestem świadomy odpowiedzialności karnej za złożenie fałszywego oświadczenia.”. Klauzula ta zastępuje pouczenie organu o odpowiedzialności karnej za składanie fałszywych oświadczeń.”,
c) ust. 9 i 9a otrzymują brzmienie:
„9. Zaistnienie okoliczności, o których mowa w ust. 8, skutkuje odwołaniem członka ze składu Rady do spraw Taryfikacji.
9a. W przypadku niezłożenia przed posiedzeniem Rady do spraw Taryfikacji oświadczenia o powiązaniach branżowych minister właściwy do spraw zdrowia odwołuje członka Rady. Jeżeli złożenie oświadczenia było niemożliwe z przyczyn technicznych lub wynikało z nieplanowanej potrzeby włączenia kolejnego członka Rady do spraw Taryfikacji do już trwającego posiedzenia, a członek Rady zgłosi do protokołu zaistnienie okoliczności określonych w ust. 8 w celu ich oceny zgodnie z ust. 12, to oświadczenie uznaje się za skutecznie złożone, pod warunkiem że wpłynie do przewodniczącego Rady do końca dnia, w którym rozpoczęło się dane posiedzenie.”,
d) po ust. 9a dodaje się ust. 9b w brzmieniu:
„9b. W przypadku poświadczenia nieprawdy w oświadczeniu o powiązaniach branżowych minister właściwy do spraw zdrowia odwołuje członka Rady do spraw Taryfikacji.”,
e) ust. 10 otrzymuje brzmienie:
„10. Osoby niebędące członkami Rady do spraw Taryfikacji, którym zleca się przygotowanie ekspertyzy pisemnej lub ustnej lub innego opracowania dla Rady - albo w związku z przygotowywaniem ekspertyzy, rekomendacji lub opinii dla Prezesa Agencji, albo w związku z przygotowywaniem stanowiska lub opinii dla Rady - składają każdorazowo przed przyjęciem zlecenia oświadczenie o powiązaniach branżowych dla każdej przygotowanej ekspertyzy, rekomendacji, stanowiska lub opinii.”,
f) po ust. 10 dodaje się ust. 10a i 10b w brzmieniu:
„10a. Prezes Agencji analizuje oświadczenia o powiązaniach branżowych złożone przez osoby, o których mowa w ust. 10, kierując się zasadą równego traktowania osób składających oświadczenia oraz biorąc pod uwagę rodzaj zgłoszonego powiązania branżowego, jego charakter i okoliczności zgłoszonego powiązania branżowego, a także ewentualny wpływ zadeklarowanego powiązania branżowego na postępowanie prowadzone przez Radę do spraw Taryfikacji. Analiza jest wykonywana w oparciu o sposób i tryb określone w przepisach wydanych na podstawie art. 31lb ust. 7c, w celu zapewnienia obiektywności rozstrzygnięć podejmowanych przez Agencję, efektywności prowadzonych przez nią postępowań oraz niedopuszczania do udziału w tych postępowaniach osób, w stosunku do których ujawnił się konflikt interesów budzący uzasadnione wątpliwości co do ich bezstronności.
10b. Prezes Agencji na podstawie dokonanej analizy, o której mowa w ust. 10a, uwzględnia ekspertyzy pisemne lub ustne lub inne opracowania dla Rady do spraw Taryfikacji lub Agencji albo ich nie uwzględnia.”,
g) ust. 11 otrzymuje brzmienie:
„11. W posiedzeniach Rady do spraw Taryfikacji mogą brać udział eksperci oraz inne osoby zaproszone przez przewodniczącego Rady, bez prawa głosu. Osoby te, przed udziałem w posiedzeniu Rady, składają oświadczenie o powiązaniach branżowych.”,
h) po ust. 11 dodaje się ust. 11a i 11b w brzmieniu:
„11a. Przewodniczący lub wiceprzewodniczący Rady do spraw Taryfikacji analizuje oświadczenia o powiązaniach branżowych złożone przez osoby, o których mowa w ust. 11, kierując się zasadą równego traktowania osób składających oświadczenia oraz biorąc pod uwagę rodzaj zgłoszonego powiązania branżowego, jego charakter i okoliczności zgłoszonego powiązania branżowego, a także ewentualny wpływ zadeklarowanego powiązania branżowego na postępowanie prowadzone przez Radę do spraw Taryfikacji. Analiza jest wykonywana w oparciu o sposób i tryb określone w przepisach wydanych na podstawie art. 31lb ust. 7c, w celu zapewnienia obiektywności rozstrzygnięć podejmowanych przez Radę, efektywności prowadzonych przez nią postępowań oraz niedopuszczania do udziału w tych postępowaniach osób, w stosunku do których ujawnił się konflikt interesów budzący wątpliwości co do ich bezstronności.
11b. Przewodniczący lub wiceprzewodniczący Rady do spraw Taryfikacji na podstawie dokonanej analizy, o której mowa w ust. 11a, dopuszcza albo nie dopuszcza do udziału w posiedzeniu Rady ekspertów oraz inne osoby przez niego zaproszone.”,
i) ust. 12 otrzymuje brzmienie:
„12. W przypadku ujawnienia konfliktu interesów członka Rady do spraw Taryfikacji prowadzący posiedzenie Rady wyłącza tego członka z udziału w pracach tej Rady oraz informuje o tym Prezesa Agencji.”,
j) po ust. 12 dodaje się ust. 12a w brzmieniu:
„12a. W przypadku zaistnienia okoliczności, o których mowa w ust. 8 pkt 4, niezależnych od członka Rady do spraw Taryfikacji, niezwłocznie informuje on Prezesa Agencji o tym fakcie. Prezes Agencji zawiesza członka w pracach Rady do spraw Taryfikacji i wyznacza mu termin usunięcia zaistniałych okoliczności, a także informuje o tym fakcie ministra właściwego do spraw zdrowia. Po bezskutecznym upływie wyznaczonego terminu minister właściwy do spraw zdrowia odwołuje członka Rady do spraw Taryfikacji.”,
k) ust. 13 otrzymuje brzmienie:
„13. Oświadczenia o powiązaniach branżowych składane przez kandydatów na członków oraz członków Rady do spraw Taryfikacji weryfikuje Centralne Biuro Antykorupcyjne.”,
l) po ust. 13 dodaje się ust. 13a w brzmieniu:
„13a. Oświadczenia o powiązaniach branżowych składane przez osoby inne niż określone w ust. 13 weryfikuje Centralne Biuro Antykorupcyjne z własnej inicjatywy lub na pisemny, uzasadniony wniosek Prezesa Agencji.”,
m) dodaje się ust. 19 w brzmieniu:
„19. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, wzór oświadczenia o powiązaniach branżowych, mając na uwadze zapewnienie przejrzystości i komunikatywności tych oświadczeń oraz potrzebę ujednolicenia ich formy.”;
7) w art. 43a:
a) w ust. 1b dodaje się zdanie drugie w brzmieniu:
„Dostęp do danych osobowych lub jednostkowych danych medycznych dotyczących ilości oraz rodzaju przepisanych świadczeniobiorcy, o którym mowa w ust. 1, leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych umożliwiający dokonanie weryfikacji, o której mowa w zdaniu pierwszym, nie wymaga zgody, o której mowa w art. 35 ust. 1a ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia.”,
b) w ust. 2 uchyla się pkt 3,
c) w ust. 3 skreśla się wyrazy „pkt 2 i 3”;
8) w art. 43b:
a) ust. 1 otrzymuje brzmienie:
„1. Świadczeniobiorcom w okresie ciąży albo połogu przysługuje bezpłatne zaopatrzenie w leki lub wyroby medyczne określone w wykazie, o którym mowa w art. 37 ust. 1 ustawy o refundacji, ustalonym w sposób określony w ust. 6.”,
b) ust. 3 i 4 otrzymują brzmienie:
„3. Podstawą bezpłatnego wydania leku lub wyrobu medycznego z apteki lub punktu aptecznego świadczeniobiorcom, o których mowa w ust. 1, jest recepta:
1) wystawiona przez:
a) lekarza lub położną, o których mowa w ust. 2, lub
b) innego lekarza - na podstawie zaświadczenia wystawionego przez lekarza lub położną, o których mowa w ust. 2;
2) zrealizowana w terminie miesiąca od dnia jej wystawienia albo od oznaczonej na recepcie daty realizacji „od dnia”.
4. Zaświadczenie, o którym mowa w ust. 3 pkt 1 lit. b, jest ważne przez okres ciąży i połogu, jednak nie dłużej niż przez 6 tygodni po upływie 15 dni od planowanej daty porodu wskazanej w tym zaświadczeniu.”,
c) w ust. 5 dodaje się zdanie drugie w brzmieniu:
„Dostęp do danych osobowych lub jednostkowych danych medycznych dotyczących ilości oraz rodzaju przepisanych świadczeniobiorcy, o którym mowa w ust. 1, leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych umożliwiający dokonanie weryfikacji, o której mowa w zdaniu pierwszym, nie wymaga zgody, o której mowa w art. 35 ust. 1a ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia.”,
d) ust. 6 i 7 otrzymują brzmienie:
„6. Minister właściwy do spraw zdrowia wskazuje spośród leków i wyrobów medycznych określonych w wykazie, o którym mowa w art. 37 ust. 1 ustawy o refundacji, leki i wyroby medyczne, które są wydawane z apteki lub punktu aptecznego bezpłatnie świadczeniobiorcom, o których mowa w ust. 1, mając na względzie:
1) konieczność zaspokojenia potrzeb zdrowotnych tych świadczeniobiorców, które wynikają z ciąży albo połogu;
2) dostępność i bezpieczeństwo stosowania w poszczególnych okresach ciąży albo okresie połogu leków i wyrobów medycznych.
7. Leki i wyroby medyczne przysługujące bezpłatnie świadczeniobiorcom, o których mowa w ust. 1, są finansowane przez Fundusz w części stanowiącej odpłatność świadczeniobiorcy, o której mowa w art. 6 ust. 2 ustawy o refundacji, obejmującej kwotę do wysokości limitu finansowania oraz dopłatę w wysokości różnicy między ceną detaliczną danego leku albo wyrobu medycznego a wysokością limitu jego finansowania.”;
9) w art. 50 ust. 23 otrzymuje brzmienie:
„23. Prezes Funduszu nie wszczyna postępowania, jeżeli wysokość kosztów świadczeń opieki zdrowotnej udzielonych w przypadkach określonych w ust. 16, które Fundusz poniósł zgodnie z ust. 15, na rzecz tej samej osoby nie przekracza kwoty 500 zł.”;
10) w art. 61j ust. 11 otrzymuje brzmienie:
„11. Prezes Funduszu może zarządzić:
1) przerwę w kontroli,
2) przedłużenie czasu trwania kontroli na czas niezbędny do przeprowadzenia czynności kontrolnych, określając termin tego przedłużenia,
3) odstąpienie od kontroli z przyczyn niezależnych od Funduszu,
4) rozszerzenie zakresu przedmiotowego kontroli
- o czym niezwłocznie powiadamia podmiot kontrolowany.”;
11) w art. 97:
a) w ust. 3:
- pkt 2e otrzymuje brzmienie:
„2e) finansowanie leków i wyrobów medycznych przysługujących świadczeniobiorcom, o których mowa w art. 43b ust. 1;”,
- pkt 3d otrzymuje brzmienie:
„3d) finansowanie zakupu szczepionek, o których mowa w art. 18 ust. 5 ustawy z dnia 5 grudnia 2008 r. o zapobieganiu oraz zwalczaniu zakażeń i chorób zakaźnych u ludzi oraz wskazanych w obwieszczeniu, o którym mowa w art. 18 ust. 5d tej ustawy, oraz produktów leczniczych, o których mowa w art. 18a ust. 2 tej ustawy;”,
- po pkt 3e dodaje się pkt 3f w brzmieniu:
„3f) finansowanie wykonywania przez apteki ogólnodostępne zadań, o których mowa w art. 94 ust. 13 ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne;”,
- po pkt 4c dodaje się pkt 4d w brzmieniu:
„4d) zawieranie i rozliczanie umów z podmiotami prowadzącymi apteki na realizację zalecanych szczepień ochronnych finansowanych ze środków publicznych zgodnie z art. 19 ust. 2a ustawy z dnia 5 grudnia 2008 r. o zapobieganiu oraz zwalczaniu zakażeń i chorób zakaźnych u ludzi;”,
b) po ust. 3h dodaje się ust. 3i-3l w brzmieniu:
„3i. Nabycie szczepionek oraz produktów leczniczych związanych z realizacją zadania, o którym mowa w ust. 3 pkt 3d, jest finansowane przez Fundusz przez przekazanie środków finansowych, w wysokości i terminach określonych przez ministra właściwego do spraw zdrowia, na wyodrębniony rachunek bankowy jednostki budżetowej podległej ministrowi właściwemu do spraw zdrowia.
3j. Jednostka, o której mowa w ust. 3i, może zaciągać zobowiązania niezbędne do nabycia szczepionek oraz produktów leczniczych, o których mowa w ust. 3i, w tym w roku poprzedzającym to nabycie, do wysokości określonej przez ministra właściwego do spraw zdrowia.
3k. Zadanie, o którym mowa w ust. 3 pkt 3e, w zakresie zakupu produktów leczniczych i wyrobów medycznych, jest finansowane przez Fundusz przez przekazanie środków finansowych, w wysokości i terminach określonych przez ministra właściwego do spraw zdrowia, na wyodrębniony rachunek bankowy jednostek budżetowych właściwych w sprawach realizacji programów polityki zdrowotnej opracowanych, realizowanych i wdrażanych przez ministra właściwego do spraw zdrowia.
3l. Jednostki, o których mowa w ust. 3k, mogą zaciągać zobowiązania niezbędne do nabycia produktów leczniczych i wyrobów medycznych, o których mowa w ust. 3k, w tym w roku poprzedzającym to nabycie, do wysokości określonej przez ministra właściwego do spraw zdrowia.”;
12) w art. 102 w ust. 5 pkt 26b otrzymuje brzmienie:
„26b) przekazywanie ministrowi właściwemu do spraw zdrowia comiesięcznych zestawień ilości zrefundowanych opakowań jednostkowych leków oraz jednostkowych wyrobów medycznych, wraz z podaniem numeru GTIN zgodnego z systemem GS1 lub innego kodu jednoznacznie identyfikującego wyrób medyczny, wydawanych na podstawie art. 43b ust. 1, w terminie do 20. dnia miesiąca następującego po miesiącu, którego dotyczy zestawienie;”;
13) w art. 116 w ust. 1 pkt 5 otrzymuje brzmienie:
„5) dotacje, w tym dotacje celowe przeznaczone na finansowanie zadań, o których mowa w art. 97 ust. 3 pkt 2f, 2g, 2h i 4c;”;
14) w art. 117 w ust. 1:
a) po pkt 3c dodaje się pkt 3d w brzmieniu:
„3d) koszt zadania, o którym mowa w art. 97 ust. 3 pkt 3f;”,
b) po pkt 4a dodaje się pkt 4aa w brzmieniu:
„4aa) koszt zadania, o którym mowa w art. 97 ust. 3 pkt 4d;”;
15) w art. 159:
a) w ust. 1 w pkt 3 kropkę zastępuje się średnikiem i dodaje się pkt 4 w brzmieniu:
„4) do umów, o których mowa w art. 97 ust. 3 pkt 4d.”,
b) po ust. 2c dodaje się ust. 2d w brzmieniu:
„2d. Umowy, o których mowa w art. 97 ust. 3 pkt 4d, są zawierane na podstawie wniosku złożonego do właściwego oddziału wojewódzkiego Funduszu przez podmioty prowadzące apteki.”,
c) ust. 3 otrzymuje brzmienie:
„3. Prezes Funduszu zamieszcza na stronie internetowej Funduszu informacje o warunkach zawarcia umowy o udzielanie świadczeń opieki zdrowotnej, o których mowa w ust. 2 i 2d.”;
16) art. 160 otrzymuje brzmienie:
„Art. 160. Świadczeniodawcy, który zawarł umowę o udzielanie świadczeń opieki zdrowotnej, przysługuje zażalenie na czynności Prezesa Funduszu lub dyrektora oddziału wojewódzkiego Funduszu dotyczące realizacji umowy, z wyłączeniem czynności Prezesa Funduszu, o których mowa w art. 61d ust. 12 i art. 61t ust. 3.”.
Art. 5. [Ustawa o zapobieganiu oraz zwalczaniu zakażeń i chorób zakaźnych u ludzi]
1) w art. 17:
a) po ust. 9 dodaje się ust. 9a-9c w brzmieniu:
„9a. Szczepienia obowiązkowe przeprowadza się zgodnie ze schematem szczepienia określonym dla danego szczepienia w przepisach wydanych na podstawie ust. 10.
9b. Lekarz sprawujący profilaktyczną opiekę zdrowotną nad osobą małoletnią przekazuje państwowemu powiatowemu inspektorowi sanitarnemu, właściwemu dla miejsca udzielania świadczeń opieki zdrowotnej, imienny wykaz:
1) osób małoletnich objętych obowiązkowymi szczepieniami ochronnymi, które nie były poddane lekarskiemu badaniu kwalifikacyjnemu albo u których nie zostały przeprowadzone obowiązkowe szczepienia ochronne, mimo niestwierdzenia u nich przeciwwskazań do szczepienia,
2) osób sprawujących prawną pieczę nad osobą małoletnią, o której mowa w pkt 1, o ile jest to możliwe do ustalenia
- zwany dalej „raportem o przypadkach niewykonania obowiązkowych szczepień ochronnych”.
9c. Raport o przypadkach niewykonania obowiązkowych szczepień ochronnych zawiera dane:
1) osoby małoletniej, o której mowa w ust. 9b pkt 1:
a) imię i nazwisko,
b) datę urodzenia,
c) numer PESEL, a w przypadku jego braku - rodzaj i numer dokumentu potwierdzającego tożsamość,
d) adres miejsca zamieszkania,
e) rodzaj i liczbę niewykonanych obowiązkowych szczepień ochronnych wraz ze wskazaniem przyczyn niewykonania tych szczepień - o ile są znane;
2) osób, o których mowa w ust. 9b pkt 2:
a) imię i nazwisko,
b) numer PESEL, a w przypadku jego braku - rodzaj i numer dokumentu potwierdzającego tożsamość,
c) adres miejsca zamieszkania.”,
b) w ust. 10:
- w pkt 2 wyrazy „do nałożenia obowiązku szczepień ochronnych na te osoby” zastępuje się wyrazami „powstania obowiązku szczepień ochronnych”,
- po pkt 2 dodaje się pkt 2a w brzmieniu:
„2a) schemat szczepienia przeciw chorobie zakaźnej obejmujący liczbę dawek i terminy ich podania wymagane dla danego szczepienia uwzględniające wiek osoby objętej obowiązkiem szczepienia,”,
- w pkt 8 na końcu dodaje się przecinek i dodaje się pkt 9 w brzmieniu:
„9) papierową lub elektroniczną formę raportu o przypadkach niewykonania obowiązkowych szczepień ochronnych oraz terminy i sposób jego przekazywania”;
2) w art. 18:
a) ust. 5c i 5d otrzymują brzmienie:
„5c. Źródłem finansowania szczepionek, o których mowa w ust. 5a, są środki pozostające w dyspozycji ministra właściwego do spraw zdrowia, w tym pochodzące z Funduszu Medycznego, o których mowa w art. 7 pkt 3a ustawy z dnia 7 października 2020 r. o Funduszu Medycznym (Dz. U. z 2023 r. poz. 1758 i 1831), oraz środki Narodowego Funduszu Zdrowia.
5d. Minister właściwy do spraw zdrowia ogłasza, w drodze obwieszczenia, wykaz zalecanych szczepień ochronnych, ze wskazaniem źródła finansowania, dla których zakup szczepionek został objęty finansowaniem, o którym mowa w ust. 5a.”,
b) po ust. 5d dodaje się ust. 5e w brzmieniu:
„5e. Wykonanie zalecanych szczepień ochronnych, o których mowa w ust. 5a, finansuje Narodowy Fundusz Zdrowia.”;
3) w art. 19:
a) po ust. 2 dodaje się ust. 2a i 2b w brzmieniu:
„2a. Przeprowadzenie u pacjenta w aptece szczepienia ochronnego na podstawie art. 86 ust. 8a ustawy z dnia 6 września 2001 r. - Prawo farmaceutyczne, a w przypadku wykonania w tej aptece, przez zatrudnionego w niej farmaceutę, badania kwalifikacyjnego, o którym mowa w ust. 5a, poprzedzającego przeprowadzenie szczepienia - również wykonanie tego badania, podlegają finansowaniu ze środków Funduszu.
2b. Minister właściwy do spraw zdrowia ogłasza, w drodze obwieszczenia, szczepienia ochronne przeprowadzane w aptece, które mogą być w całości lub w części finansowane ze środków publicznych.”,
b) ust. 5-5b otrzymują brzmienie:
„5. Jeżeli ubezpieczony wykona zalecane szczepienie ochronne lub lekarskie badanie kwalifikacyjne u innego świadczeniodawcy niż wskazany w ust. 2, lub w aptece, ponosi koszty badania i przeprowadzenia szczepienia.
5a. Wykonanie zalecanego szczepienia ochronnego oraz szczepienia przeciw COVID-19 jest poprzedzone badaniem kwalifikacyjnym w celu wykluczenia przeciwwskazań do wykonania tego szczepienia, które może w każdym przypadku przeprowadzić lekarz lub felczer, a w przypadku badania osoby dorosłej, również:
1) lekarz dentysta, pielęgniarka, położna, ratownik medyczny;
2) fizjoterapeuta, diagnosta laboratoryjny lub farmaceuta, który uzyskał dokument potwierdzający ukończenie szkolenia teoretycznego, dostępnego na platformie e-learningowej Centrum Medycznego Kształcenia Podyplomowego.
5b. Zalecane szczepienie ochronne oraz szczepienie przeciw COVID-19 przeprowadza w przypadku:
1) osoby dorosłej - lekarz, lekarz dentysta, felczer, pielęgniarka, położna, higienistka szkolna, ratownik medyczny, fizjoterapeuta, diagnosta laboratoryjny albo farmaceuta,
2) osoby do ukończenia 18. roku życia - lekarz, felczer, pielęgniarka, położna, higienistka szkolna lub ratownik medyczny
- posiadający kwalifikacje określone w przepisach wydanych na podstawie art. 17 ust. 10 pkt 3 albo przepisach wydanych na podstawie art. 76 pkt 1 ustawy z dnia 10 grudnia 2020 r. o zawodzie farmaceuty (Dz. U. z 2022 r. poz. 1873 oraz z 2023 r. poz. 1234) lub którzy ukończyli kurs teoretyczny i praktyczny do szczepień, którego program został zatwierdzony przez Centrum Medycznego Kształcenia Podyplomowego.”,
c) po ust. 5b dodaje się ust. 5ba w brzmieniu:
„5ba. Farmaceuta przeprowadza zalecane szczepienie ochronne oraz szczepienie przeciw COVID-19 w przypadkach określonych w ust. 5b, jeżeli:
1) samodzielnie dokonał kwalifikacji, o której mowa w ust. 5a, w stosunku do pacjenta podlegającemu zaszczepieniu albo
2) kwalifikacji do szczepienia dokonał lekarz, felczer, lekarz dentysta i wystawił on receptę na szczepionkę.”,
d) ust. 6 i 7 otrzymują brzmienie:
„6. Do przeprowadzania zalecanych szczepień ochronnych oraz szczepień przeciw COVID-19 stosuje się odpowiednio przepisy art. 17 ust. 3, z tym że w przypadku gdy badanie kwalifikacyjne dotyczy szczepienia zalecanego, nie stosuje się art. 17 ust. 5.
7. Osoby przeprowadzające zalecane szczepienie ochronne dokonują wpisów potwierdzających wykonanie szczepienia w Karcie Szczepień oraz:
1) informują osobę poddawaną szczepieniu o możliwości wydania na jej koszt Międzynarodowej Książeczki Szczepień;
2) wydają na koszt osoby szczepionej Międzynarodową Książeczkę Szczepień.”,
e) uchyla się ust. 8,
f) ust. 10 otrzymuje brzmienie:
„10. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia:
1) wykaz zalecanych szczepień ochronnych,
2) wzór Międzynarodowej Książeczki Szczepień oraz sposób jej wydawania i dokonywania w niej wpisów
- uwzględniając zalecenia Światowej Organizacji Zdrowia i cele nadzoru epidemiologicznego.”;
4) w art. 21a:
a) ust. 2 otrzymuje brzmienie:
„2. Po przeprowadzonym badaniu kwalifikacyjnym osoba je przeprowadzająca dokonuje wpisu o tym badaniu do Karty Szczepień ze wskazaniem daty i godziny przeprowadzonego badania.”,
b) dodaje się ust. 5 w brzmieniu:
„5. Jednostka podległa ministrowi właściwemu do spraw zdrowia właściwa w zakresie systemów informacyjnych ochrony zdrowia udostępnia dane o szczepieniach Narodowemu Funduszowi Zdrowia w celu realizacji rozliczeń udzielonych świadczeń opieki zdrowotnej.”.
Art. 6. [Ustawa o wyrobach medycznych]
1) w art. 58:
a) uchyla się ust. 2,
b) ust. 3 otrzymuje brzmienie:
„3. Nadzór nad reklamą wyrobów medycznych jest sprawowany przez Prezesa Urzędu.”;
2) w art. 104 ust. 2-4 otrzymują brzmienie:
„2. Organ może odstąpić od wymierzenia kar pieniężnych, o których mowa w niniejszym rozdziale, jeżeli zdarzenie miało charakter incydentalny, nie stwarzało ryzyka, a podmiot, najpóźniej w terminie wskazanym przez Prezesa Urzędu, przy prowadzeniu czynności, w trakcie których stwierdzono uchybienie prawa, podjął działania w celu jego usunięcia i poinformował Prezesa Urzędu o podjęciu tych działań.
3. Kary pieniężne, o których mowa w art. 74-103, nakłada, w drodze decyzji administracyjnej, Prezes Urzędu.
4. Kary pieniężne są uiszczane na rachunek bankowy Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych.”.
Art. 7. [Przedłużenie okresu obowiązywania decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego]
Art. 8. [Stosowanie przepisów dotychczasowych]
Art. 9. [Podjęcie postępowań zawieszonych na wniosek strony]
2. W przypadku niezłożenia wniosku, o którym mowa w ust. 1, w wyznaczonym terminie postępowanie umarza się.
Art. 10. [Postępowania omówione w art. 81 ust. 1 pkt 2 lit. b oraz art. 103 ust. 1 pkt 2 Prawa farmaceutycznego]
Art. 11. [Zespoły koordynacyjne]
2. W terminie 3 miesięcy od dnia wejścia w życie niniejszej ustawy Prezes Narodowego Funduszu Zdrowia dostosuje działanie zespołów koordynacyjnych, o których mowa w ust. 1, do wymogów określonych w art. 16b ust. 4-6 i 8 ustawy zmienianej w art. 1.
3. Prezes Narodowego Funduszu Zdrowia w terminie 30 dni od dnia wejścia w życie niniejszej ustawy dostosuje regulaminy zespołów koordynacyjnych, o których mowa w ust. 1, obowiązujące przed dniem wejścia w życie niniejszej ustawy do przepisów ustawy zmienianej w art. 1, w brzmieniu nadanym niniejsza ustawą.
Art. 12. [Składanie deklaracji o braku konfliktu interesów przez członków Komisji Ekonomicznej]
Art. 13. [Kadencje członków Rady Przejrzystości]
2. Z dniem 9 marca 2024 r. minister właściwy do spraw zdrowia powołuje członków Rady Przejrzystości na nową wspólną kadencję.
Art. 14. [Składanie deklaracji o powiązaniach branżowych przez członków Rady Przejrzystości]
2. Przepis ust. 1 stosuje się do członków Rady do spraw Taryfikacji, o której mowa w art. 31sa ust. 1 ustawy zmienianej w art. 4, w odniesieniu do składanych przez nich oświadczeń o powiązaniach branżowych, o których mowa w art. 31sa ust. 8a ustawy zmienianej w art. 4.
3. Deklaracje o braku konfliktu interesów, o których mowa w art. 31s ust. 9 ustawy zmienianej w art. 4, oraz oświadczenia o braku konfliktu interesów, o których mowa w art. 31sa ust. 9 ustawy zmienianej w art. 4, złożone przed dniem wejścia w życie niniejszej ustawy odpowiednio przez członków Rady Przejrzystości lub członków Rady do spraw Taryfikacji, są weryfikowane przez Centralne Biuro Antykorupcyjne na podstawie przepisów dotychczasowych.
4. Deklaracje o braku konfliktu interesów, o których mowa w art. 31s ust. 9 ustawy zmienianej w art. 4, oraz oświadczenia o braku konfliktu interesów, o których mowa w art. 31sa ust. 9 ustawy zmienianej w art. 4, złożone przed dniem wejścia w życie niniejszej ustawy przez osoby niebędące odpowiednio członkami Rady Przejrzystości i Rady do spraw Taryfikacji zachowują ważność w zakresie ekspertyz, innych opracowań lub uwag, jakich dotyczyły.
Art. 15. [Informacja o uruchomieniu możliwości dostępu do danych osobowych lub jednostkowych danych medycznych]
Art. 16. [Ważność recept]
1) na lek, na podstawie art. 43b ust. 3 ustawy zmienianej w art. 4,
2) na produkt leczniczy, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny w liczbie wynikającej z art. 96a ust. 3 albo 5 ustawy zmienianej w art. 2 w brzmieniu dotychczasowym,
3) na produkt leczniczy, środek spożywczy specjalnego przeznaczenia żywieniowego lub wyrób medyczny, w ilości określonej w art. 96a ust. 2 albo 4 ustawy zmienianej w art. 2 w brzmieniu dotychczasowym
- zachowują ważność i mogą być realizowane na zasadach dotychczasowych.
2. Recepty wystawione przed dniem wejścia w życie niniejszej ustawy w postaci papierowej, na których zostały przepisane produkty lecznicze o:
1) kategorii dostępności, o której mowa w art. 23a ust. 1 pkt 4 ustawy zmienianej w art. 2, albo
2) innej kategorii dostępności, niż określona w pkt 1, zawierające w swoim składzie substancję psychotropową lub środek odurzający, z których którakolwiek substancja lub środek należy do grupy II-N, III-P albo IV-P określonej w ustawie z dnia 29 lipca 2005 r. o przeciwdziałaniu narkomanii (Dz. U. z 2023 r. poz. 172 oraz z 2022 r. poz. 2600)
- zachowują ważność i mogą być realizowane na zasadach dotychczasowych.
3. Zaświadczenia, o których mowa w art. 43b ust. 3 pkt 2 ustawy zmienianej w art. 4, wystawione przed dniem wejścia w życie niniejszej ustawy, zachowują ważność przez okres określony w art. 43b ust. 4 ustawy zmienianej w art. 4, w brzmieniu nadanym niniejszą ustawą.
Art. 17. [Postępowanie o objęcie refundacją leku]
2. W przypadku postępowania o objęcie refundacją leku wszczętego i niezakończonego przed dniem wejścia w życie niniejszej ustawy, gdy wnioskodawca w rozumieniu art. 2 pkt 27 ustawy zmienianej w art. 1 nie wyznaczył przedstawiciela podmiotu odpowiedzialnego w rozumieniu art. 2 pkt 35a ustawy zmienianej w art. 2, w brzmieniu nadanym niniejszą ustawą, postępowanie to zawiesza się do czasu wyznaczenia tego przedstawiciela, pod rygorem pozostawienia sprawy bez rozpoznania, po bezskutecznym upływie 3 miesięcy od dnia wejścia w życie niniejszej ustawy.
Art. 18. [Urzędowa marża detaliczna]
| od | do | zasada marży |
| - | 10,00 zł | 0,35 zł + 35% * x |
| 10,01 zł | 20,00 zł | 3,85 zł + 25% * (x - 10,00 zł) |
| 20,01 zł | 40,00 zł | 6,35 zł + 15% * (x - 20,00 zł) |
| 40,01 zł | 80,00 zł | 9,35 zł + 10% * (x - 40,00 zł) |
| 80,01 zł | 160,00 zł | 13,35 zł + 5% * (x - 80,00 zł) |
| 160,01 zł | 640,00 zł | 17,35 zł + 2,75% * (x - 160,00 zł) |
| 640,01 zł | | 30,55 zł + 2% * (x - 640,00 zł) |
Art. 19. [Uchwały w sprawie rozkładu godzin pracy aptek ogólnodostępnych]
2. Do dnia wejścia w życie uchwał, o których mowa w art. 94 ust. 3 ustawy zmienianej w art. 2, w brzmieniu nadanym niniejszą ustawą, w aptece ogólnodostępnej prowadzącej na terenie danego powiatu ekspedycję w porze nocnej może być pobierana opłata za tę ekspedycję w wysokości określonej w przepisach wydanych na podstawie art. 94 ust. 3 ustawy zmienianej w art. 2, w brzmieniu dotychczasowym.
3. W 2024 r. podmiot prowadzący aptekę ogólnodostępną przekazuje zarządowi powiatu albo prezydentowi miasta na prawach powiatu, w którym znajduje się ta apteka, rozkład godzin pracy tej apteki na dany rok w terminie do dnia 31 stycznia 2024 r.
4. W 2024 r. uchwała zarządu powiatu oraz zarządzenie prezydenta miasta na prawach powiatu w sprawie dyżurów w porze nocnej lub dyżurów w dni wolne od pracy są wydawane na okres do dnia 31 grudnia 2024 r.
Art. 20. [Termin ogłoszenia leków lub wyrobów medycznych przysługujących świadczeniobiorcom omówionym w art. 43b ust. 1 ustawy o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych]
Art. 21. [Przetargi]
Art. 22. [Limity finansowania]
2. Do czasu rozpoczęcia obowiązywania limitów finansowania za jednostkę surowca farmaceutycznego służącego do sporządzenia leku recepturowego, ustalonych przez Prezesa Narodowego Funduszu Zdrowia w pierwszym wykazie, o którym mowa w ust. 1, wydawanie leku recepturowego przygotowywanego z surowców farmaceutycznych lub z leków gotowych, dla których została wydana decyzja administracyjna o objęciu refundacją, odbywa się na zasadach dotychczasowych wynikających z art. 6 ust. 5 ustawy zmienianej w art. 1.
3. Pierwszy wykaz, o którym mowa w ust. 1, będzie obowiązywał po upływie miesiąca od dnia jego ustalenia.
Art. 23. [Wejście w życie]
1) art. 2 pkt 12 lit. c, który wchodzi w życie z dniem następującym pod dniu ogłoszenia, z mocą od dnia 18 sierpnia 2023 r.;
2) art. 4 pkt 11 lit. b, który wchodzi w życie z dniem następującym po dniu ogłoszenia;
3) art. 4 pkt 11 lit. a tiret drugie i czwarte oraz art. 5, które wchodzą w życie z dniem 1 października 2023 r.;
4) art. 1 pkt 35 lit. d i pkt 41 lit. f, art. 2 pkt 1 lit. a, pkt 2, 10, 15 i 16 oraz art. 4 pkt 11 lit. a tiret trzecie i pkt 14, które wchodzą w życie z dniem 1 stycznia 2024 r.;
5) art. 2 pkt 12 lit. d w zakresie art. 96a ust. 7ab, który wchodzi w życie z dniem 1 marca 2024 r.;
6) art. 1 pkt 35 lit. a-c, które wchodzą w życie z dniem 1 lipca 2024 r.;
7) art. 1 pkt 6 lit. b, który wchodzi w życie z dniem 1 stycznia 2025 r.
1) Niniejszą ustawą zmienia się ustawy: ustawę z dnia 6 września 2001 r. - Prawo farmaceutyczne, ustawę z dnia 12 marca 2004 r. o pomocy społecznej, ustawę z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych, ustawę z dnia 5 grudnia 2008 r. o zapobieganiu oraz zwalczaniu zakażeń i chorób zakaźnych u ludzi oraz ustawę z dnia 7 kwietnia 2022 r. o wyrobach medycznych.
2) Zmiany wymienionego rozporządzenia zostały ogłoszone w Dz. Urz. UE L 87 z 31.03.2009, str. 174, Dz. Urz. UE L 348 z 31.12.2010, str. 1 oraz Dz. Urz. UE L 198 z 25.07.2019, str. 241.
3) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2022 r. poz. 1726, 1855, 2339 i 2600 oraz z 2023 r. poz. 289, 818, 852, 1234, 1834 i 1860.
4) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2022 r. poz. 2674 i 2770 oraz z 2023 r. poz. 605, 650, 658, 1234, 1429, 1675, 1692, 1733, 1831 i 1872.
5) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2022 r. poz. 2674 i 2770 oraz z 2023 r. poz. 605, 650, 658, 1234, 1429, 1675, 1692, 1733, 1831 i 1872.
[1] Art. 1 pkt 24 lit. a) w brzmieniu ustalonym przez obwieszczenie Marszałka Sejmu Rzeczypospolitej Polskiej z dnia 2 października 2023 r. o sprostowaniu błędu (Dz.U. poz. 2105). Zmiana weszła w życie 3 października 2023 r.
- Data ogłoszenia: 2023-09-20
- Data wejścia w życie: 2023-11-01
- Data obowiązywania: 2023-11-01

- USTAWA z dnia 7 kwietnia 2022 r. o wyrobach medycznych
- USTAWA z dnia 6 września 2001 r. Prawo farmaceutyczne
- USTAWA z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych
- USTAWA z dnia 12 maja 2011 r. o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych
- USTAWA z dnia 12 marca 2004 r. o pomocy społecznej
- USTAWA z dnia 5 grudnia 2008 r. o zapobieganiu oraz zwalczaniu zakażeń i chorób zakaźnych u ludzi
- USTAWA z dnia 12 maja 2011 r. o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych
REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA