REKLAMA
Dziennik Ustaw - rok 2021 poz. 1707
ROZPORZĄDZENIE
MINISTRA KLIMATU I ŚRODOWISKA1)
z dnia 9 sierpnia 2021 r.
w sprawie wymagań jakościowych dla biokomponentów, metod badań jakości biokomponentów oraz sposobu pobierania próbek biokomponentów2)
Na podstawie art. 22 ust. 6 ustawy z dnia 25 sierpnia 2006 r. o biokomponentach i biopaliwach ciekłych (Dz. U. z 2021 r. poz. 1355 i 1642) zarządza się, co następuje:
§ 1. [Zakres regulacji]
1) wymagania jakościowe dla biokomponentów:
a) bioetanolu (etanolu jako komponentu benzyny silnikowej), stanowiące załącznik nr 1 do rozporządzenia,
b) estru metylowego, stanowiące załącznik nr 2 do rozporządzenia,
c) biowęglowodorów ciekłych przeznaczonych do wytwarzania paliw ciekłych lub biopaliw ciekłych stosowanych do silników z zapłonem iskrowym, stanowiące załącznik nr 3 do rozporządzenia,
d) biowęglowodorów ciekłych przeznaczonych do wytwarzania paliw ciekłych lub biopaliw ciekłych stosowanych do silników z zapłonem samoczynnym, stanowiące załącznik nr 4 do rozporządzenia,
e) bio propanu-butanu, stanowiące załącznik nr 5 do rozporządzenia;
2) metody badań jakości biokomponentów, stanowiące załącznik nr 6 do rozporządzenia;
3) sposób pobierania próbek biokomponentów, stanowiący załącznik nr 7 do rozporządzenia.
§ 2. [Wyłączenie stosowania wymagań jakościowych dla biokomponentów]
§ 3. [Wejście w życie]
Minister Klimatu i Środowiska: M. Kurtyka
1) Minister Klimatu i Środowiska kieruje działem administracji rządowej - energia, na podstawie § 1 ust. 2 pkt 1 rozporządzenia Prezesa Rady Ministrów z dnia 6 października 2020 r. w sprawie szczegółowego zakresu działania Ministra Klimatu i Środowiska (Dz. U. z 2021 r. poz. 941).
2) Niniejsze rozporządzenie zostało notyfikowane Komisji Europejskiej w dniu 30 kwietnia 2021 r. pod numerem 2021/0267/PL, zgodnie z § 4 rozporządzenia Rady Ministrów z dnia 23 grudnia 2002 r. w sprawie sposobu funkcjonowania krajowego systemu notyfikacji norm i aktów prawnych (Dz. U. poz. 2039 oraz z 2004 r. poz. 597), które wdraża dyrektywę (UE) 2015/1535 Parlamentu Europejskiego i Rady z dnia 9 września 2015 r. ustanawiającą procedurę udzielania informacji w dziedzinie przepisów technicznych oraz zasad dotyczących usług społeczeństwa informacyjnego (ujednolicenie) (Dz. Urz. UE L 241 z 17.09.2015, str. 1).
3) Niniejsze rozporządzenie było poprzedzone rozporządzeniem Ministra Gospodarki z dnia 17 grudnia 2010 r. w sprawie wymagań jakościowych dla biokomponentów, metod badań jakości biokomponentów oraz sposobu pobierania próbek biokomponentów (Dz. U. z 2017 r. poz. 506), które traci moc z dniem wejścia w życie niniejszego rozporządzenia zgodnie z art. 15 ustawy z dnia 19 lipca 2019 r. o zmianie ustawy o biokomponentach i biopaliwach ciekłych oraz niektórych innych ustaw (Dz. U. poz. 1527).
Załączniki do rozporządzenia Ministra Klimatu i Środowiska
z dnia 9 sierpnia 2021 r. (poz. 1707)
Załącznik nr 1
WYMAGANIA JAKOŚCIOWE DLA BIOETANOLU (ETANOLU JAKO KOMPONENTU BENZYNY SILNIKOWEJ)1)
| Właściwość | Jednostka | Zakresy | |
| minimum | maksimum | ||
| Zawartość etanolu i alkoholi wyższych nasyconych3) | % (m/m) | 98,7 | - |
| Zawartość mono alkoholi wyższych nasyconych (C3 – C5)2), 3) | % (m/m) | - | 2,0 |
| Zawartość metanolu3) | % (m/m) | - | 1,0 |
| Zawartość wody | % (m/m) | - | 0,300 |
| Zawartość nieorganicznych chlorków | mg/kg | - | 6,0 |
| Zawartość miedzi | mg/kg | - | 0,100 |
| Całkowita kwasowość (wyrażona jako zawartość kwasu octowego) | % (m/m) | - | 0,007 |
| Wygląd |
| przejrzysty i bezbarwny | |
| Zawartość fosforu | mg/l | - | 0,15 |
| Zawartość suchej pozostałości po odparowaniu | mg/100 ml | - | 10 |
| Zawartość siarki | mg/kg | - | 10,0 |
| Przewodność elektryczna | μS/cm | - | 2,5 |
| Zawartość siarczanów | mg/kg | - | 4,0 |
| 1) Wymagania odnoszą się do bioetanolu nieskażonego. 2) W przypadku przeznaczenia bioetanolu do syntezy eterów zawartość alkoholi wyższych jest uzgadniana między dostawcą a odbiorcą. 3) Wynik badania odnosi się do próbki niezawierającej wody. Opis metody badania zawarto w załączniku nr 6 w pkt 4. | |||
Załącznik nr 2
WYMAGANIA JAKOŚCIOWE DLA ESTRU METYLOWEGO
| Właściwość | Jednostka | Zakresy | ||
| minimum | maksimum | |||
| Zawartość estru metylowego kwasów tłuszczowych (FAME)1) | % (m/m) | 96,5 | - | |
| Gęstość w temperaturze 15°C | kg/m³ | 860 | 900 | |
| Lepkość w temperaturze 40°C | mm²/s | 3,50 | 5,00 | |
| Temperatura zapłonu | °C | 101 | - | |
| Zawartość siarki | mg/kg | - | 10,0 | |
| Liczba cetanowa |
| 51,0 | - | |
| Zawartość popiołu siarczanowego | % (m/m) | - | 0,02 | |
| Zawartość wody | mg/kg | - | 500 | |
| Zawartość zanieczyszczeń stałych | mg/kg | - | 24 | |
| Badanie działania korodującego na miedzi (3 h w temperaturze 50°C) | stopień korozji | stopień korozji 1 | ||
| Stabilność oksydacyjna w temperaturze 110°C | h | 8,0 | - | |
| Liczba kwasowa | mg KOH/g | - | 0,50 | |
| Liczba jodowa | g jodu/100g | - | 120 | |
| Zawartość estru metylowego kwasu linolenowego | % (m/m) | - | 12,0 | |
| Zawartość estrów metylowych wielonienasyconych kwasów tłuszczowych (≥4 wiązania podwójne) | % (m/m) | - | 1,00 | |
| Zawartość metanolu | % (m/m) | - | 0,20 | |
| Zawartość monoacylogliceroli | % (m/m) | - | 0,70 | |
| Zawartość diacylogliceroli | % (m/m) | - | 0,20 | |
| Zawartość triacylogliceroli | % (m/m) | - | 0,20 | |
| Zawartość wolnego glicerolu | % (m/m) | - | 0,02 | |
| Zawartość ogólnego glicerolu | % (m/m) | - | 0,25 | |
| Zawartość metali grupy I (Na + K) | mg/kg | - | 5,0 | |
| Zawartość metali grupy II (Ca + Mg) | mg/kg | - | 5,0 | |
| Zawartość fosforu | mg/kg | - | 4,0 | |
| Temperatura zablokowania zimnego filtru, CFPP | °C | - | 02) | -103) |
| Temperatura mętnienia | °C | - | 52) | -33) |
| 1) Nie dopuszcza się innych składników oprócz dodatków uszlachetniających. Jeżeli w FAME występują w sposób naturalny estry C17, fakt ten może wpływać na zaniżenie oznaczonej zawartości estrów metylowych kwasów tłuszczowych. W takim przypadku w celu weryfikacji wykonuje się badanie referencyjne zgodnie z procedurą [4], podaną w normie PN-EN 14214. 2) Dla okresu letniego trwającego od 16 kwietnia do 30 września. 3) Dla okresu przejściowego oraz okresu zimowego trwających od 1 października do 15 kwietnia. | ||||
Załącznik nr 3
WYMAGANIA JAKOŚCIOWE DLA BIOWĘGLOWODORÓW CIEKŁYCH PRZEZNACZONYCH DO WYTWARZANIA PALIW CIEKŁYCH LUB BIOPALIW CIEKŁYCH STOSOWANYCH DO SILNIKÓW Z ZAPŁONEM ISKROWYM
| Właściwość | Jednostka | Zakresy | |
| minimum | maksimum | ||
| Zawartość siarki | mg/kg | - | 10,0 |
| Okres indukcyjny | min | 360 | - |
| Zawartość żywic obecnych (po przemyciu rozpuszczalnikiem) | mg/100 cm³ | - | 5 |
| Badanie działania korodującego na miedzi (3 h w temperaturze 50°C) | stopień korozji | klasa 1 | |
| Skład frakcyjny: |
|
|
|
| – temperatura końca destylacji | °C | - | 210 |
| – pozostałość po destylacji | % (V/V) | - | 2 |
| Zawartość benzenu | % (V/V) | - | 1,0 |
| Zawartość zanieczyszczeń | mg/kg | - | 10,0 |
| Całkowita zawartość chlorowców | mg/kg | - | 2,0 |
| Zawartość fosforu | mg/l | - | 1,0 |
Załącznik nr 4
WYMAGANIA JAKOŚCIOWE DLA BIOWĘGLOWODORÓW CIEKŁYCH PRZEZNACZONYCH DO WYTWARZANIA PALIW CIEKŁYCH LUB BIOPALIW CIEKŁYCH STOSOWANYCH DO SILNIKÓW Z ZAPŁONEM SAMOCZYNNYM
| Właściwość | Jednostka | Zakresy | |
| minimum | maksimum | ||
| Gęstość (w temperaturze 15°C) | kg/m³ | - | 845,0 |
| Zawartość siarki | mg/kg | - | 10,0 |
| Zawartość wielopierścieniowych węglowodorów aromatycznych | % (m/m) | - | 8,0 |
| Temperatura zapłonu | °C | wyższa niż 55 |
|
| Zawartość zanieczyszczeń | mg/kg | - | 15 |
| Badanie działania korodującego na miedzi (3 h w temperaturze 50°C) | stopień korozji | klasa 1 | |
| Skład frakcyjny: 95% (V/V) destyluje do temperatury | °C | - | 360 |
| Liczba kwasowa | mg KOH/g | - | 0,2 |
| Liczba bromowa | g Br/100 g | - | 1,0 |
| Całkowita zawartość chlorowców | mg/kg | - | 5,0 |
| Zawartość fosforu | mg/kg | - | 3,0 |
| Zawartość krzemu | mg/kg | - | 3,0 |
| Zawartość sodu | mg/kg | - | 3,0 |
| Zawartość potasu | mg/kg | - | 3,0 |
| Zawartość cynku | mg/kg | - | 0,3 |
| Zawartość miedzi | mg/kg | - | 0,3 |
Załącznik nr 5
WYMAGANIA JAKOŚCIOWE DLA BIO PROPANU-BUTANU
| Właściwość | Jednostka | Zakresy | |
| minimum | maksimum | ||
| Całkowita zawartość dienów | % (m/m) | - | 0,5 |
| Zawartość 1,3-butadienu | % (m/m) | - | 0,10 |
| Siarkowodór |
| brak | |
| Całkowita zawartość siarki (po wprowadzeniu substancji zapachowej) | mg/kg | - | 30 |
| Badanie działania korodującego na płytce miedzianej (1 h w temperaturze 40°C) | ocena | klasa 1 | |
| Pozostałość po odparowaniu | mg/kg | - | 60 |
| Prężność par, oszacowana w temperaturze 40°C | kPa | - | 1550 |
| Zawartość wody |
| nie wykryto | |
| Zawartość węglowodorów C3-C4 | % (m/m) | 94,5 | - |
Załącznik nr 6
METODY BADAŃ JAKOŚCI BIOKOMPONENTÓW
I. Metody badań jakości bioetanolu, w zakresie poszczególnych parametrów tego biokomponentu.
1. Zawartość etanolu i alkoholi wyższych nasyconych oznacza się metodą chromatografii gazowej, polegającą na wprowadzeniu porcji próbki do chromatografu gazowego (GC), wydzieleniu etanolu i alkoholi wyższych z pozostałych składników, wykryciu ich przy pomocy detektora płomieniowo-jonizacyjnego (FID) i określeniu ich zawartości w odniesieniu do wewnętrznego wzorca na podstawie współczynnika odpowiedzi.
1.1. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej wyposażenie, sposób obliczenia, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15721. W obliczeniu zawartości etanoli i wyższych alkoholi należy uwzględnić zawartość wody w próbce.
2. Zawartość mono alkoholi wyższych nasyconych (C3 – C5) oznacza się metodą chromatografii gazowej, polegającą na wprowadzeniu porcji próbki do chromatografu gazowego (GC), wydzieleniu mono alkoholi wyższych nasyconych (C3 – C5) z pozostałych składników, wykryciu ich przy pomocy detektora płomieniowo-jonizacyjnego (FID) i określeniu ich zawartości w odniesieniu do wewnętrznego wzorca na podstawie współczynnika odpowiedzi.
2.1. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej wyposażenie, sposób obliczenia, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15721.
3. Zawartość metanolu oznacza się metodą chromatografii gazowej, polegającą na wprowadzeniu porcji próbki do chromatografu gazowego (GC), wydzieleniu metanolu z pozostałych składników, wykryciu go przy pomocy detektora płomieniowo-jonizacyjnego (FID) i określeniu jego zawartości w odniesieniu do wewnętrznego wzorca na podstawie współczynnika odpowiedzi.
3.1. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej wyposażenie, sposób obliczenia, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15721.
4. W celu odniesienia uzyskanych metodą GC wg PN-EN 15721 wyników zawartości etanolu i wyższych alkoholi, zawartości wyższych nasyconych (C3-C5) mono alkoholi oraz zawartości metanolu do próbki niezawierającej wody należy uwzględnić zawartość wody w próbce poprzez jej odrębne oznaczenie odpowiednią metodą, po czym – w oparciu o wynik oznaczenia wody Cwody, wyrażony w % (m/m) – obliczenie współczynnika korekcyjnego W, pozwalającego odnieść wyniki uzyskane dla próbki z wodą do próbki niezawierającej wody, wg wzoru:
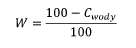
wynik oznaczenia metodą GC zawartości etanoli i wyższych alkoholi, zawartości wyższych nasyconych (C3-C5) mono alkoholi oraz zawartości metanolu należy przemnożyć przez współczynnik korekcyjny W i podać w raporcie jako wynik badania skorygowany dla próbki niezawierającej wody.
5. Zawartość wody oznacza się metodą:
1) miareczkowania kulometrycznego Karla Fischera, polegającą na wprowadzeniu zważonej próbki do naczynia do miareczkowania aparatu kulometrycznego Karla Fischera, w którym jod do reakcji Karla Fischera wydziela się elektrolitycznie na anodzie, proporcjonalnie do ilości wody zawartej w próbce, albo
2) miareczkowania potencjometrycznego Karla Fishera, polegającą na wprowadzeniu zważonej próbki do naczynia do miareczkowania aparatu potencjometrycznego Karla Fischera, w którym obecna woda jest miareczkowana z zastosowaniem odczynnika Karla Fishera.
5.1. W przypadku oznaczania zawartości wody w sposób określony w pkt 5 ppkt 1, gdy cała woda zostaje odmiareczkowana, nadmiar jodu wykrywany jest przez czujnik elektrometrycznego punktu końcowego i miareczkowanie zostaje przerwane. Ze stechiometrii reakcji wynika, że jeden mol jodu reaguje z jednym molem wody, stąd ilość wody jest proporcjonalna do całkowitego ładunku, zgodnie z prawem Faradaya.
5.2. W przypadku oznaczania zawartości wody w sposób określony w pkt 5 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie próbki i aparatury, test kontrolny aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15489.
5.3. W przypadku oznaczania zawartości wody w sposób określony w pkt 5 ppkt 2 jod do reakcji jest wprowadzany w obecności ditlenku siarki, metanolu i odpowiedniej zasady azotowej. Na podstawie stechiometrii reakcji jeden mol jodu reaguje z jednym molem wody.
5.4. W przypadku oznaczania zawartości wody w sposób określony w pkt 5 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie próbki i aparatury, test kontrolny aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15692.
6. Zawartość nieorganicznych chlorków oznacza się metodą:
1) potencjometryczną, polegającą na odparowaniu odważonej ilości próbki w łaźni wodnej, rozpuszczeniu suchej pozostałości w dejonizowanej wodzie i określeniu zawartości nieorganicznych chlorków w drodze miareczkowania potencjometrycznego, albo
2) chromatografii jonowej, polegającą na odparowaniu próbki w łaźni wodnej, rozpuszczeniu suchej pozostałości w wodzie i określeniu zawartości chlorków przez porównanie na chromatografie powierzchni pików badanej próbki ze standardową krzywą kalibracji.
6.1. W przypadku wykonania oznaczenia w sposób określony w pkt 6 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej przygotowanie, kalibrację, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15484.
6.2. W przypadku wykonania oznaczenia w sposób określony w pkt 6 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, przygotowanie roztworu kalibracyjnego i aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15492.
7. Zawartość miedzi oznacza się metodą:
1) atomowej spektrometrii absorpcyjnej z atomizacją elektrotermiczną w piecu grafitowym, polegającą na wprowadzaniu porcji próbki na powierzchnię wewnętrzną lub półeczkę kuwety oraz ogrzewaniu kuwety zgodnie z odpowiednim programem temperaturowym, albo
2) optycznej spektrometrii emisyjnej indukcyjnie sprzężonej plazmy, polegającą na wprowadzeniu próbki do komory mgielnej spektrometru emisyjnego indukcyjnie sprzężonej plazmy.
7.1. W przypadku oznaczania zawartości miedzi w sposób określony w pkt 7 ppkt 1 ilość światła zaabsorbowanego przez atomy miedzi w czasie ostatniego etapu programu jest mierzona w określonych jednostkach czasu, a zintegrowana absorbancja Aj, wytworzona przez miedź zawarta w próbce jest porównywana z krzywą wzorcową wyznaczoną na podstawie sygnałów roztworów wzorcowych miedzi w etanolu.
7.2. W przypadku oznaczania zawartości miedzi w sposób określony w pkt 7 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie roztworu do próby ślepej i roztworów wzorcowych, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15488.
7.3. W przypadku oznaczania zawartości miedzi w sposób określony w pkt 7 ppkt 2 zawartość miedzi oznacza się poprzez porównanie emisji pierwiastka w próbce analitycznej z emisją w roztworach wzorcowych przy tej samej długości fali.
7.4. W przypadku oznaczania zawartości miedzi w sposób określony w pkt 7 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie roztworu do próby ślepej i roztworów wzorcowych, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15837.
8. Całkowitą kwasowość (wyrażoną jako zawartość kwasu octowego) oznacza się metodą miareczkowania kolorymetrycznego, polegającą na zmieszaniu próbki z taką samą porcją wody niezawierającą ditlenku węgla i zmiareczkowaniu roztworem wodorotlenku potasu w obecności fenoloftaleiny do momentu zobojętnienia związków o charakterze kwaśnym.
8.1. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, aparaturę, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badań określa norma PN-EN 15491.
9. Wygląd sprawdza się metodą wizualną, polegającą na ocenie koloru oraz przezroczystości, przez porównanie z próbką wody na tle białym oraz czarnym.
9.1. Sposób wykonania oznaczenia, stosowaną aparaturę, sposób interpretacji i podawania wyników, a także sporządzanie sprawozdania z badania określa norma PN-EN 15769.
10. Zawartość fosforu oznacza się metodą:
1) spektrometryczną, polegającą na odparowaniu próbki, rozpuszczeniu suchej pozostałości w wodzie i dodaniu kwaśnego roztworu zawierającego jony molibdenu i antymonu w celu uzyskania kompleksu antymonowo-fosforowo-molibdenowego, albo
2) optycznej spektrometrii emisyjnej indukcyjnie sprzężonej plazmy, polegającą na wprowadzeniu próbki do komory mgielnej spektrometru emisyjnego indukcyjnie sprzężonej plazmy.
10.1. W przypadku oznaczania zawartości fosforu w sposób określony w pkt 10 ppkt 1 kompleks jest poddawany działaniu kwasu askorbinowego w celu uzyskania kompleksu molibdenowego o mocnej niebieskiej barwie. Zawartość fosforu uzyskuje się poprzez pomiar absorbancji kompleksu przy długości fali równej 880 nm.
10.2. W przypadku oznaczania zawartości fosforu w sposób określony w pkt 10 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki i materiały, aparaturę, kalibrację, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badań określa norma PN-EN 15487.
10.3. W przypadku oznaczania zawartości fosforu w sposób określony w pkt 10 ppkt 2 zawartość fosforu oznacza się poprzez porównanie emisji pierwiastka w próbce analitycznej z emisją w roztworach wzorcowych przy tej samej długości fali.
10.4. W przypadku oznaczania zawartości fosforu w sposób określony w pkt 10 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie roztworu do próby ślepej i roztworów wzorcowych, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15837.
11. Zawartość suchej pozostałości po odparowaniu oznacza się metodą grawimetryczną, polegającą na zważeniu pozostałości po odparowaniu alkoholu w łaźni wodnej i osuszeniu w suszarce.
11.1. Sposób wykonania oznaczenia, rodzaj aparatury i wyposażenie, sposób obliczenia oraz podawania wyników, a także precyzję metody określa norma PN-EN 15691.
12. Zawartość siarki oznacza się metodą:
1) rentgenowskiej spektroskopii fluorescencyjnej z dyspersją fali, polegającą na poddaniu badanej próbki, znajdującej się w kuwecie pomiarowej, działaniu pierwotnego promieniowania o określonej długości fali, pochodzącego z lampy rentgenowskiej, albo
2) fluorescencji UV, polegającą na wykorzystaniu zjawiska fluorescencji ditlenku siarki wzbudzonego promieniowaniem ultrafioletowym, powstałego uprzednio na skutek spalenia badanej próbki w określonych warunkach, albo
3) optycznej spektrometrii emisyjnej indukcyjnie sprzężonej plazmy, polegającą na wprowadzeniu próbki do komory mgielnej spektrometru emisyjnego indukcyjnie sprzężonej plazmy.
12.1. W przypadku oznaczania zawartości siarki w sposób określony w pkt 12 ppkt 1 należy wyznaczyć zawartość siarki na podstawie krzywej kalibracji określonej dla odpowiedniego zakresu pomiarowego.
12.2. W przypadku oznaczania zawartości siarki w sposób określony w pkt 12 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie roztworów kalibracyjnych, procedurę kalibracji, sposób podawania wyników, precyzję metody, a także sposób sporządzania sprawozdania z badania określa norma PN-EN 15485.
12.3. W przypadku oznaczania zawartości siarki w sposób określony w pkt 12 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki i materiały, kalibrację i weryfikację aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sposób sporządzania sprawozdania z badania określa norma PN-EN 15486.
12.4. W przypadku oznaczania zawartości siarki w sposób określony w pkt 12 ppkt 3 zawartość siarki oznacza się poprzez porównanie emisji pierwiastka w próbce analitycznej z emisją w roztworach wzorcowych przy tej samej długości fali.
12.5. W przypadku oznaczania zawartości siarki w sposób określony w pkt 12 ppkt 3 sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie roztworu do próby ślepej i roztworów wzorcowych, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15837.
13. Przewodność elektryczną oznacza się na podstawie pomiaru przewodnictwa elektrycznego za pomocą konduktometru z zastosowaniem naczynka pomiarowego, w temperaturze próbki (25+/–0,1)°C.
13.1. Sposób wykonania oznaczenia, rodzaj stosowanej aparatury, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15938.
14. Zawartość siarczanów określa się metodą chromatografii jonowej, polegającą na odparowaniu próbki w łaźni wodnej, rozpuszczeniu suchej pozostałości w wodzie i określeniu zawartości jonów siarczanowych przez porównanie na chromatografie powierzchni pików badanej próbki z krzywą wzorcową.
14.1. Sposób wykonania oznaczenia, rodzaj stosowanej aparatury, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15492.
15. W przypadkach spornych należy stosować metodę badania wskazaną w normie PN-EN 15376.
II. Metody badań jakości estru metylowego, w zakresie poszczególnych parametrów tego biokomponentu.
1. Zawartość estru metylowego kwasów tłuszczowych oznacza się metodą chromatografii gazowej z użyciem wzorca wewnętrznego, polegającą na rozdziale mieszaniny na poszczególne składniki w fazie gazowej.
1.1. Warunki chromatograficzne powinny być tak dobrane, aby były dobrze widoczne piki estrów metylowych kwasu lignocerowego (C24) i nerwonowego (C24:1). Integracja powinna być tak przeprowadzona, aby były uwzględnione piki, począwszy od estru metylowego kwasu mirystynowego (C14) do piku estru metylowego kwasu nerwonowego (C24:1).
1.2. Zawartość estru metylowego kwasów tłuszczowych oblicza się na podstawie całkowitej powierzchni pików estrów metylowych od C14 do C24:1 oraz powierzchni piku odpowiadającego estrowi metylowemu kwasu heptadekanowego i wyraża się jako ułamek masowy określony w procentach.
1.3. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie próbki, sposób podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14103.
2. Gęstość estru metylowego w temperaturze 15°C oznacza się:
1) metodą z areometrem, polegającą na pomiarze gęstości badanej próbki o określonej temperaturze, za pomocą areometru zanurzonego w próbce znajdującej się w cylindrze, albo
2) metodą oscylacyjną, wprowadzając próbkę (o objętości około 1 ml) do celi pomiarowej gęstościomierza oscylacyjnego, termostatowanej w celu utrzymania temperatury odniesienia 15°C.
2.1. W przypadku oznaczania gęstości w temperaturze 15°C w sposób określony w pkt 2 ppkt 1 należy odczytać wskazanie na podziałce areometru, zanotować temperaturę badanej próbki i przeliczyć wynik pomiaru przy użyciu wzoru określonego w załączniku C do normy PN-EN 14214, aby odnieść go do temperatury 15°C.
2.2. W przypadku oznaczania gęstości w temperaturze 15°C w sposób określony w pkt 2 ppkt 1 sposób wykonania oznaczenia, rodzaj aparatury oraz jej przygotowanie i kontrolę, przygotowanie próbki, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 3675.
2.3. W przypadku oznaczania gęstości w temperaturze 15°C w sposób określony w pkt 2 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury i jej przygotowanie, przygotowanie próbki, kalibrację aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 12185.
3. Lepkość estru metylowego w temperaturze 40°C oznacza się metodą polegającą na pomiarze czasu przepływu określonej objętości badanej próbki, pod wpływem sił grawitacyjnych, przez wzorcowany, szklany lepkościomierz kapilarny w powtarzalnych warunkach, w określonej i precyzyjnie utrzymywanej, stałej i ściśle kontrolowanej temperaturze.
3.1. Lepkość w temperaturze 40°C oblicza się, mnożąc zmierzony czas przepływu stałej objętości cieczy, pomiędzy kreskami zbiornika pomiarowego, przez stałą lepkościomierza.
3.2. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej wzorcowanie, sposób obliczenia i podawania wyników, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 3104.
3.3. Precyzję metody oznaczania lepkości w temperaturze 40°C określa załącznik A do normy PN-EN 14214.
4. Temperaturę zapłonu estru metylowego oznacza się:
1) szybką metodą równowagową, w tyglu zamkniętym, polegającą na umieszczeniu badanej próbki w tyglu i podgrzewaniu jej do chwili zaobserwowania zapłonu par na powierzchni badanej próbki, albo
2) metodą zamkniętego tygla Pensky'ego-Martensa, polegającą na umieszczeniu badanej próbki w tyglu i podgrzewaniu, przy ciągłym mieszaniu, do chwili, gdy wprowadzone przez otwór w pokrywie tygla źródło zapłonu spowoduje zapłon par na powierzchni badanej próbki.
4.1. W przypadku oznaczania temperatury zapłonu w sposób określony w pkt 4 ppkt 1 do badania należy użyć 2 ml badanej próbki i zastosować przyrząd pomiarowy wyposażony w urządzenie rejestrujące temperaturę.
4.2. W przypadku oznaczania temperatury zapłonu w sposób określony w pkt 4 ppkt 1 najniższą temperaturę, w której następuje zapłon par na powierzchni badanej próbki, przyjmuje się jako temperaturę zapłonu w warunkach otoczenia.
4.3. W przypadku oznaczania temperatury zapłonu w sposób określony w pkt 4 ppkt 1 zmierzoną temperaturę zapłonu badanej próbki w warunkach otoczenia koryguje się do normalnego ciśnienia atmosferycznego.
4.4. W przypadku oznaczania temperatury zapłonu w sposób określony w pkt 4 ppkt 1:
1) sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej przygotowanie, sposób obliczenia i podawania wyników, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 3679;
2) precyzję metody określa załącznik A do normy PN-EN 14214.
4.5. W przypadku oznaczania temperatury zapłonu w sposób określony w pkt 4 ppkt 2 najniższą temperaturę, w której przyłożenie źródła zapłonu spowoduje zapłon par badanej próbki i szerzenie się płomienia ponad powierzchnią cieczy, przyjmuje się jako temperaturę zapłonu w warunkach otoczenia.
4.6. W przypadku oznaczania temperatury zapłonu w sposób określony w pkt 4 ppkt 2 zmierzoną temperaturę zapłonu badanej próbki w warunkach otoczenia koryguje się do normalnego ciśnienia atmosferycznego.
4.7. W przypadku oznaczania temperatury zapłonu w sposób określony w pkt 4 ppkt 2 należy stosować procedurę A oraz używać aparatury do określania temperatury zapłonu wyposażonej w odpowiednie urządzenie wykrywające (termiczne lub jonizacyjne).
4.8. W przypadku oznaczania temperatury zapłonu w sposób określony w pkt 4 ppkt 2:
1) sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej przygotowanie, postępowanie z próbką, sposób obliczenia i podawania wyników, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 2719;
2) precyzję metody określa załącznik A do normy PN-EN 14214.
5. Zawartość siarki oznacza się metodą:
1) rentgenowskiej spektrometrii fluorescencyjnej z dyspersją fali, polegającą na poddaniu badanej próbki, znajdującej się w kuwecie pomiarowej, działaniu pierwotnego promieniowania o określonej długości fali, pochodzącego z lampy rentgenowskiej, albo
2) fluorescencji w nadfiolecie, polegającą na wykorzystaniu zjawiska fluorescencji ditlenku siarki wzbudzonego promieniowaniem ultrafioletowym, powstałego uprzednio na skutek spalenia badanej próbki w określonych warunkach, albo
3) spektrometrii fluoroscencji rentgenowskiej z dyspersją energii, polegającą na umieszczeniu w strumieniu wzbudzającego promieniowania lampy rentgenowskiej próbki analitycznej znajdującej się w kuwecie dostosowanej do okna przepuszczającego promieniowanie rentgenowskie.
5.1. W przypadku oznaczania zawartości siarki w sposób określony w pkt 5 ppkt 1 należy wyznaczyć zawartość siarki na podstawie mierzonych szybkości zliczeń rentgenowskiego promieniowania fluorescencyjnego linii S-K oraz promieniowania tła, korzystając z krzywej wzorcowania.
5.2. W przypadku oznaczania zawartości siarki w sposób określony w pkt 5 ppkt 1:
1) sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury i jej przygotowanie, sposób obliczenia i podawania wyników określa norma PN-EN-ISO 20884;
2) precyzję metody określa załącznik A do normy PN-EN 14214.
5.3. W przypadku oznaczania zawartości siarki w sposób określony w pkt 5 ppkt 2 miarą zawartości siarki w badanej próbce jest intensywność fluorescencyjnego promieniowania ultrafioletowego.
5.4. W przypadku oznaczania zawartości siarki w sposób określony w pkt 5 ppkt 2:
1) sposób wykonania oznaczenia, rodzaj aparatury i jej przygotowanie, stosowane odczynniki, sposób obliczenia i podawania wyników określa norma PN-EN ISO 20846;
2) precyzję metody określa załącznik A do normy PN-EN 14214.
5.5. W przypadku oznaczania zawartości siarki w sposób, o którym mowa w pkt 5 ppkt 3, mierzy się intensywność linii K-L2,3 promieniowania rentgenowskiego charakterystycznego siarki i porównuje skumulowaną liczbę zliczeń z wartościami krzywej wzorcowej uzyskanej dla roztworów wzorcowych o zawartości siarki obejmujący badany zakres stężeń.
5.6. W przypadku oznaczania zawartości siarki w sposób, o którym mowa w pkt 5 ppkt 3, sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 13032.
6. Liczbę cetanową oznacza się metodą silnikową, polegającą na porównaniu własności samozapłonowych oleju napędowego z analogicznymi właściwościami mieszanek paliw wzorcowych o znanych liczbach cetanowych, przy zastosowaniu silnika badawczego w znormalizowanych warunkach.
6.1. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej przygotowanie, przygotowanie próbki i aparatury, kalibrację, sposób obliczenia i podawania wyników, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 5165.
6.2. Precyzję metody określa załącznik A do normy PN-EN 14214.
7. Zawartość popiołu siarczanowego oznacza się metodą wagową, polegającą na obliczeniu udziału popiołu siarczanowego, uzyskanego przez spalenie badanej próbki i reakcję pozostałości po spopieleniu z kwasem siarkowym.
7.1. Badaną próbkę spala się do chwili, gdy pozostanie tylko popiół i węgiel. Po schłodzeniu produktów spalania, poddaje się je działaniu kwasu siarkowego i prażeniu w temperaturze 775°C, aż zakończy się utlenianie węgla. Następnie popiół schładza się, ponownie poddaje działaniu kwasu siarkowego i prażeniu, aż do uzyskania stałej masy.
7.2. Sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, a także sporządzanie sprawozdania z badania określa norma PN-ISO 3987.
7.3. Precyzję metody określa załącznik A do normy PN-EN 14214.
8. Zawartość wody oznacza się metodą miareczkowania kulometrycznego, polegającą na wprowadzeniu zważonej próbki do naczynia do miareczkowania aparatu kulometrycznego Karla Fischera, w którym jod do reakcji Karla Fischera wydziela się elektrolitycznie na anodzie, proporcjonalnie do ilości wody zawartej w próbce.
8.1. Gdy cała zawartość wody zostaje odmiareczkowana, nadmiar jodu wykrywa czujnik elektrometrycznego punktu końcowego i miareczkowanie zostaje przerwane. Ze stechiometrii reakcji wynika, że jeden mol jodu reaguje z jednym molem wody, stąd ilość wody jest proporcjonalna do całkowitego ładunku, zgodnie z prawem Faradaya.
8.2. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury i jej przygotowanie oraz test kontrolny, przygotowanie próbki, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 12937.
9. Zawartość zanieczyszczeń stałych określa się metodą polegającą na oznaczeniu udziału masy zanieczyszczeń odfiltrowanych na sączku w odniesieniu do całkowitej masy próbki.
9.1. Określoną ilość przygotowanej próbki sączy się w temperaturze pokojowej, z zastosowaniem próżni, przez uprzednio zważony sączek oraz oznacza wagowo udział masy zanieczyszczeń pozostałych na sączku w odniesieniu do całkowitej masy próbki.
9.2. Sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury i jej przygotowanie, przygotowanie próbki, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 12662.
10. Działanie korodujące na miedzi określa się porównawczo w stosunku do znormalizowanych wzorców korozji.
10.1. Płytkę miedzianą zanurza się w badanej próbce o określonej objętości, a następnie ogrzewa w ściśle określonych warunkach. Po zakończeniu ogrzewania płytkę miedzianą wyjmuje się, przemywa i ocenia jej barwę, porównując z wzorcami korozji.
10.2. Sposób wykonania badania, stosowane odczynniki i materiały, rodzaj aparatury, sposób interpretacji i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 2160.
11. Stabilność oksydacyjną w temperaturze 110°C oznacza się metodą:
1) polegającą na przepuszczeniu przez badaną próbkę strumienia oczyszczonego powietrza; lotne związki, uwalniane z próbki w procesie utleniania, przechodzą wraz z powietrzem do naczynia zawierającego wodę demineralizowaną lub destylowaną, zaopatrzonego w elektrodę do pomiaru przewodności właściwej, połączoną z jednostką pomiarową wskazującą koniec okresu indukcyjnego, albo
2) przyspieszonego utleniania, polegającą na przepuszczeniu przez badaną próbkę strumienia oczyszczonego powietrza; lotne związki, uwalniane z próbki w procesie utleniania, przechodzą wraz z powietrzem do naczynia zawierającego wodę demineralizowaną lub destylowaną, zaopatrzonego w elektrodę do pomiaru przewodności właściwej, połączoną z jednostką pomiarową wskazującą koniec okresu indukcyjnego w chwili, gdy przewodność właściwa zaczyna gwałtownie wzrastać; przyspieszony wzrost jest spowodowany dysocjacją lotnych kwasów karboksylowych, które tworzą się w procesie utleniania i zostają zaabsorbowane w wodzie.
11.1. W przypadku oznaczania stabilności oksydacyjnej w sposób, o którym mowa w pkt 11 ppkt 1, sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14112.
11.2. W przypadku oznaczania stabilności oksydacyjnej w sposób, o którym mowa w pkt 11 ppkt 2, sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15751.
12. Liczbę kwasową oznacza się metodą miareczkową, polegającą na rozpuszczeniu badanej próbki w mieszaninie rozpuszczalników i miareczkowaniu rozcieńczonym roztworem wodorotlenku potasu, przy zastosowaniu fenoloftaleiny jako wskaźnika do ustalenia punktu końcowego miareczkowania.
12.1. Sposób wykonania oznaczenia, odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14104.
13. Liczbę jodową oznacza się metodą:
1) miareczkową, polegającą na rozpuszczeniu badanej próbki w mieszaninie rozpuszczalników, dodaniu odczynnika Wijsa, a następnie, po określonym czasie, dodaniu do próbki jodku potasu i wody oraz miareczkowaniu uwolnionego jodu mianowanym roztworem tiosiarczanu sodu, albo
2) obliczeniową, polegającą na wykorzystaniu składu estrów metylowych, wyrażonego w ułamku masowym w procentach, albo
3) obliczeniową na podstawie danych z chromatografii gazowej, w której jako dane wejściowe stosuje się wyniki oznaczania chromatografii gazowej dla poszczególnych estrów metylowych kwasów tłuszczowych.
13.1. W przypadku oznaczania liczby jodowej w sposób określony w pkt 13 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14111.
13.2. W przypadku oznaczania liczby jodowej w sposób określony w pkt 13 ppkt 2 sposób obliczania liczby jodowej oraz podawania wyniku określa załącznik B do normy PN-EN 14214.
13.3. W przypadku oznaczania stabilności oksydacyjnej w sposób, o którym mowa w pkt 13 ppkt 3, sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 16300.
14. Zawartość estru metylowego kwasu linolenowego oznacza się metodą chromatografii gazowej z użyciem wzorca wewnętrznego, polegającą na rozdziale mieszaniny na poszczególne składniki w fazie gazowej.
14.1. Warunki chromatograficzne powinny być tak dobrane, aby były dobrze widoczne piki estrów metylowych kwasu lignocerowego (C24) i nerwonowego (C24:1). Integracja powinna być tak przeprowadzona, aby były uwzględnione piki, począwszy od estru metylowego kwasu mirystynowego (C14) do piku estru metylowego kwasu nerwonowego (C24:1).
14.2. Zawartość estru metylowego kwasu linolenowego oblicza się na podstawie całkowitej powierzchni pików estrów metylowych od C14 do C24:1 oraz powierzchni piku odpowiadającego estrowi metylowemu kwasu heptadekanowego i wyraża się jako ułamek masowy określony w procentach.
14.3. Sposób wykonania oznaczenia, stosowane odczynniki i materiały, rodzaj aparatury, przygotowanie próbki, sposób podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14103.
15. Zawartość metanolu oznacza się metodą polegającą na ogrzewaniu próbki w temperaturze 80ºC w hermetycznie zamkniętej fiolce, a następnie po osiągnięciu stanu równowagi, nastrzykiwaniu określonej części fazy gazowej do chromatografu, gdzie metanol jest wykrywany z użyciem detektora płomieniowo-jonizacyjnego, a jego ilość jest określana w odniesieniu do wzorca zewnętrznego.
15.1. Metanol może być także oznaczany poprzez dodanie wzorca wewnętrznego do próbki, a następnie określany z użyciem współczynnika kalibracji wewnętrznej.
15.2. Sposób wykonania oznaczenia, stosowane odczynniki i roztwory wzorcowe, rodzaj aparatury, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14110.
16. Zawartość monoacylogliceroli, diacylogliceroli, triacylogliceroli oraz ogólnego glicerolu oznacza się metodą polegającą na analizie pochodnych silanowych metodą chromatografii gazowej na krótkiej kolumnie kapilarnej z cienkowarstwowym filmem, z zastosowaniem bezpośredniego dozowania na kolumnę oraz detektora płomieniowo-jonizacyjnego.
16.1. Po przeprowadzeniu kalibracji analizę ilościową wykonuje się metodą wzorca wewnętrznego, a całkowitą (ogólną) zawartość glicerolu oblicza się na podstawie uzyskanych wyników.
16.2. Sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14105.
17. Zawartość wolnego glicerolu oznacza się metodą polegającą na:
1) analizie pochodnych silanowych metodą chromatografii gazowej na krótkiej kolumnie kapilarnej z cienkowarstwowym filmem, z zastosowaniem bezpośredniego dozowania na kolumnę oraz detektora płomieniowo-jonizacyjnego, albo
2) dodaniu, do badanej próbki, etanolu, wody, heksanu i wzorca wewnętrznego, co spowoduje utworzenie dwóch faz i ilościowe przeniesienie wolnego glicerolu do fazy dolnej, której analiza, metodą chromatografii gazowej, pozwala na ilościowe oznaczenie stężenia wolnego glicerolu.
17.1. W przypadku oznaczania zawartości wolnego glicerolu w sposób określony w pkt 17 ppkt 1, po przeprowadzeniu kalibracji, analizę ilościową wolnego glicerolu wykonuje się metodą wzorca wewnętrznego.
17.2. W przypadku oznaczania zawartości wolnego glicerolu w sposób określony w pkt 17 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14105.
17.3. W przypadku oznaczania zawartości wolnego glicerolu w sposób określony w pkt 17 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14106.
18. Zawartość sodu oznacza się:
1) bezpośrednio metodą atomowej spektrometrii absorpcyjnej przy długości fali równej 589,0 nm, rozpuszczając uprzednio badaną próbkę w roztworze ksylenu, albo
2) metodą optycznej emisyjnej analizy spektralnej z plazmą wzbudzoną indukcyjnie, rozcieńczając uprzednio badaną próbkę frakcją naftową.
18.1. W przypadku oznaczania zawartości sodu w sposób określony w pkt 18 ppkt 1 stosowane roztwory wzorcowe sporządza się z organicznego związku sodu w postaci soli, rozpuszczonego w mieszaninie ksylenu i oleju do rozcieńczeń.
18.2. W przypadku oznaczania zawartości sodu w sposób określony w pkt 18 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób podawania wyników, a także precyzję metody i sporządzanie sprawozdania z badania określa norma PN-EN 14108.
18.3. W przypadku oznaczania zawartości sodu w sposób określony w pkt 18 ppkt 2 zawartość sodu określa się przez porównanie intensywności emisji atomowej roztworu wzorcowego i próbki przy określonych długościach fal.
18.4. W przypadku oznaczania zawartości sodu w sposób określony w pkt 18 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki i roztwory wzorcowe, rodzaj aparatury i jej przygotowanie, sposób obliczania i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14538.
19. Zawartość potasu oznacza się:
1) bezpośrednio metodą atomowej spektrometrii absorpcyjnej przy długości fali równej 766,5 nm, rozpuszczając uprzednio badaną próbkę w roztworze ksylenu i stabilizatora, albo
2) metodą optycznej emisyjnej analizy spektralnej z plazmą wzbudzoną indukcyjnie, rozcieńczając uprzednio badaną próbkę frakcją naftową.
19.1. W przypadku oznaczania zawartości potasu w sposób określony w pkt 19 ppkt 1 stosowane roztwory wzorcowe sporządza się z organicznego związku potasu w postaci soli, rozpuszczonego w mieszaninie ksylenu i stabilizatora.
19.2. W przypadku oznaczania zawartości potasu w sposób określony w pkt 19 ppkt 1 sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14109.
19.3. W przypadku oznaczania zawartości potasu w sposób określony w pkt 19 ppkt 2 zawartość potasu określa się przez porównanie intensywności emisji atomowej roztworu wzorcowego i próbki przy określonych długościach fal.
19.4. W przypadku oznaczania zawartości potasu w sposób określony w pkt 19 ppkt 2 sposób wykonania oznaczenia, stosowane odczynniki i roztwory wzorcowe, rodzaj aparatury i jej przygotowanie, sposób obliczania i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14538.
19.5. Precyzję oznaczania łącznej zawartości sodu i potasu, w przypadku oznaczania zawartości sodu zgodnie z normą PN-EN 14108 oraz potasu zgodnie z normą PN-EN 14109, określa załącznik A do normy PN-EN 14214.
20. Zawartość wapnia i magnezu oznacza się metodą optycznej emisyjnej analizy spektralnej z plazmą wzbudzoną indukcyjnie, rozcieńczając uprzednio badaną próbkę frakcją naftową.
20.1. Zawartość wapnia i magnezu określa się przez porównanie intensywności emisji atomowej roztworu wzorcowego i próbki przy określonych długościach fal.
20.2. Sposób wykonania oznaczenia, stosowane odczynniki i roztwory wzorcowe, rodzaj aparatury i jej przygotowanie, sposób obliczania i podawania wyników, precyzję, a także sporządzanie sprawozdania z badania określa norma PN-EN 14538.
21. Zawartość fosforu oznacza się metodą:
1) polegającą na rozpuszczeniu badanej próbki w ksylenie i wprowadzeniu, w formie aerozolu, wraz z roztworami wzorcowymi przygotowanymi z organicznego związku fosforu, do plazmy argonowej sprzężonej indukcyjnie, albo
2) optycznej spektometrii emisyjnej plazmy wzbudzonej indukcyjnie, polegającą na przepuszczeniu przez spektrometr próbki rozpuszczonej w nafcie.
21.1. W przypadku oznaczania zawartości fosforu w sposób, o którym mowa w pkt 21 ppkt 1, zawartość fosforu oznacza się przez porównanie emisji tego pierwiastka w roztworze badanej próbki z emisją wzorców przy tej samej długości fali.
21.2. W przypadku oznaczania zawartości fosforu w sposób, o którym mowa w pkt 21 ppkt 1, sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 14107.
21.3. W przypadku oznaczania zawartości fosforu w sposób, o którym mowa w pkt 21 ppkt 2, zawartość fosforu oznacza się poprzez porównanie z roztworem wzorcowym.
21.4. W przypadku oznaczania zawartości fosforu w sposób, o którym mowa w pkt 21 ppkt 2, sposób wykonania oznaczenia, stosowane odczynniki, rodzaj aparatury, sposób obliczenia i podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 16294.
22. Zawartość estrów metylowych wielonienasyconych kwasów tłuszczowych (>4 wiązania podwójne) oznacza się metodą chromatografii gazowej z wykorzystaniem wzorca wewnętrznego estru metylowego C23:0.
22.1. Sposób wykonania oznaczenia, rodzaj stosowanej aparatury, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 15779.
23. Temperaturę zablokowania zimnego filtru (CFPP) oznacza się metodą:
1) polegającą na zasysaniu badanej próbki przez znormalizowany filtr do pipety w warunkach kontrolowanego podciśnienia i w temperaturze obniżanej co 1°C, do chwili zatrzymania lub spowolnienia przepływu, tak że czas napełniania pipety przekroczy 60 sekund lub paliwo nie spływa całkowicie do naczynia pomiarowego, albo
2) polegającą na zasysaniu badanej próbki przez znormalizowany filtr siatkowy do pipety w warunkach kontrolowanego podciśnienia 2 kPa i w temperaturze obniżanej co 1°C z liniowym przebiegiem chłodzenia łaźni, do chwili zatrzymania lub spowolnienia przepływu, tak że czas napełniania pipety przekroczy 60 sekund lub paliwo nie spływa całkowicie do naczynia pomiarowego.
23.1. W przypadku oznaczania temperatury zablokowania zimnego filtru w sposób określony w pkt 23 ppkt 1 sposób wykonania oznaczenia, rodzaj stosowanej aparatury, przygotowanie próbki, sposób podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 116.
23.2. W przypadku oznaczania temperatury zablokowania zimnego filtru w sposób określony w pkt 23 ppkt 2 sposób wykonania oznaczenia, rodzaj stosowanej aparatury, przygotowanie próbki, sposób podawania wyników, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN 16329.
24. Temperaturę mętnienia oznacza się metodą polegającą na pomiarze temperatury badanej próbki oziębianej z określoną szybkością w łaźni oziębiającej i obserwacji wyglądu tej próbki.
24.1. Temperaturę, w której obserwuje się zmętnienie przy dnie badanej próbki, przyjmuje się jako temperaturę mętnienia tej próbki.
24.2. Sposób wykonania oznaczenia, rodzaj stosowanej aparatury, precyzję metody, a także sporządzanie sprawozdania z badania określa norma PN-EN ISO 3015.
25. W przypadkach spornych należy stosować metodę badania wskazaną w normie PN-EN 14214.
III. Metody badań jakości biowęglowodorów ciekłych przeznaczonych do wytwarzania paliw ciekłych lub biopaliw ciekłych stosowanych do silników z zapłonem iskrowym.
1. Zawartość siarki, okres indukcyjny, zawartość żywic obecnych, działanie korodujące na miedzi, skład frakcyjny oraz zawartość benzenu oznacza się metodami badań tych parametrów dla benzyn silnikowych określonymi w przepisach wydanych na podstawie art. 26 pkt 1 ustawy z dnia 25 sierpnia 2006 r. o systemie monitorowania i kontrolowania jakości paliw (Dz. U. z 2021 r. poz. 133, 694, 1093 i 1642).
2. Zawartość zanieczyszczeń określa się metodą grawimetryczną poprzez przefiltrowanie znanej objętości paliwa przez uprzednio zważony filtr membranowy.
2.1. Masa zanieczyszczenia cząsteczkowego jest określona poprzez wzrost masy filtra membranowego względem filtra kontrolnego.
2.2. Sposób wykonania badania, stosowane odczynniki i materiały, rodzaj aparatury, sposób pobierania i przygotowania próbek, sposób przygotowania aparatury, sposób wykonania oznaczenia, obliczenia i podawanie wyników, precyzję, a także sporządzanie protokołu z badania określa norma ASTM D 5452.
3. Całkowitą zawartość chlorowców oznacza się metodą mikrokulometrii oksydacyjnej, polegającą na wstrzykiwaniu próbki do ogrzewanej strefy rury pirolizera, w której jest odparowywana. Produkty spalania wraz z chlorowcowodorami są przepuszczane w formie rozproszonej przez płuczkę z kwasem siarkowym, wprowadzane do naczynka kulometrycznego i miareczkowane generowanymi elektrolitycznie jonami srebra.
3.1. Wskazania mikrokulometru sprawdza się z zastosowaniem ciekłych związków organicznych.
3.2. Sposób wykonania badania, stosowane odczynniki i materiały, rodzaj aparatury, sposób pobierania i przygotowania próbek, sposób przygotowania aparatury, sposób wykonania oznaczenia, obliczenia i podawanie wyników, precyzję, a także sporządzanie protokołu z badania określa norma PN-EN 14077.
4. Zawartość fosforu oznacza się poprzez spalenie materii organicznej w próbce w obecności tlenku cynku. Pozostałość rozpuszcza się w kwasie siarkowym i poddaje się reakcji z molibdenianem amonu i siarczanem hydrazyny.
4.1. Absorbancja kompleksu błękitu molibdenowego jest proporcjonalna do zawartości fosforu w próbce i jest mierzona w około 820 nm w kuwecie o długości 5 cm.
4.2. Sposób wykonania badania, stosowane odczynniki i materiały, rodzaj aparatury, sposób pobierania i przygotowania próbek, sposób przygotowania aparatury, sposób wykonania oznaczenia, obliczenia i podawanie wyników, precyzję, a także sporządzanie protokołu z badania określa norma ASTM D 3231.
IV. Metody badań jakości biowęglowodorów ciekłych przeznaczonych do wytwarzania paliw ciekłych lub biopaliw ciekłych stosowanych do silników z zapłonem samoczynnym.
1. Gęstość, zawartość siarki, zawartość wielopierścieniowych węglowodorów aromatycznych, temperaturę zapłonu, zawartość zanieczyszczeń, działanie korodujące na miedzi oraz skład frakcyjny (95% V/V destyluje do temperatury) oznacza się metodami badań tych parametrów dla oleju napędowego określonymi w przepisach wydanych na podstawie art. 26 pkt 1 ustawy z dnia 25 sierpnia 2006 r. o systemie monitorowania i kontrolowania jakości paliw.
2. Liczbę kwasową oznacza się metodą badania tego parametru dla estru metylowego stanowiącego samoistne paliwo określoną w przepisach wydanych na podstawie art. 26 pkt 2 ustawy z dnia 25 sierpnia 2006 r. o systemie monitorowania i kontrolowania jakości paliw.
3. Liczbę bromową oznacza się metodą elektrometryczną polegającą na rozpuszczeniu określonej ilości badanej próbki w rozpuszczalniku w temperaturze 0°C do 5°C i miareczkowaniu roztworem bromku-bromianu.
3.1. Punkt końcowy miareczkowania jest wskazany przez nagłą zmianę potencjału elektrometrycznego na aparaturze.
3.2. Sposób wykonania badania, rodzaj aparatury, sposób wykonania oznaczenia, obliczenia i podawanie wyników, precyzję, a także sporządzanie protokołu z badania określa norma ISO 3839.
4. Całkowitą zawartość chlorowców oznacza się metodą mikrokulometrii oksydacyjnej, polegającą na wstrzykiwaniu próbki do ogrzewanej strefy rury pirolizera, w której jest odparowywana. Produkty spalania wraz z chlorowcowodorami są przepuszczane w formie rozproszonej przez płuczkę z kwasem siarkowym, wprowadzane do naczynka kulometrycznego i miareczkowane generowanymi elektrolitycznie jonami srebra.
4.1. Wskazania mikrokulometru sprawdza się z zastosowaniem ciekłych związków organicznych.
4.2. Sposób wykonania badania, stosowane odczynniki i materiały, rodzaj aparatury, sposób pobierania i przygotowania próbek, sposób przygotowania aparatury, sposób wykonania oznaczenia, obliczenia i podawanie wyników, precyzję, a także sporządzanie protokołu z badania określa norma PN-EN 14077.
5. Zawartość krzemu oznacza się metodą optycznej spektrometrii emisyjnej ze wzbudzaniem w plazmie indukowanej, polegającą na wprowadzeniu do spektrometru roztworu wzorcowego z wewnętrznym wzorcem oraz badanej próbki z wewnętrznym wzorcem i porównaniu uzyskanych wyników do wzorca wewnętrznego.
5.1. Sposób wykonania badania, rodzaj aparatury, sposób wykonania oznaczenia, obliczenia i podawanie wyników, precyzję, a także sporządzanie protokołu badania określa norma ASTM D 7111.
6. Zawartość fosforu, sodu, potasu, cynku i miedzi oznacza się metodą optycznej spektrometrii emisyjnej ze wzbudzaniem w plazmie indukowanej polegającą na rozpuszczeniu określonej ilości próbki w rozpuszczalniku organicznym i wprowadzeniu tego roztworu do spektrometru.
6.1. Zawartość pierwiastków oblicza się poprzez porównanie do stężeń wzorcowych.
6.2. Sposób wykonania badania, rodzaj aparatury, sposób wykonania oznaczenia, obliczenia i podawanie wyników, precyzję, a także sporządzanie protokołu badania określa norma PN-EN 16476.
V. Metody badań jakości bio propanu-butanu.
1. Całkowitą zawartość dienów, obecność siarkowodoru, całkowitą zawartość siarki, działanie korodujące na miedzi, pozostałość po odparowaniu, prężność par, oszacowaną w temperaturze 40°C oraz zawartość wody oznacza się metodami badań tych parametrów dla gazu skroplonego (LPG) określonymi w przepisach wydanych na podstawie art. 26 pkt 3 ustawy z dnia 25 sierpnia 2006 r. o systemie monitorowania i kontrolowania jakości paliw.
2. Zawartość węglowodorów C3-C4 określa się metodą chromatografii gazowej, polegającą na wprowadzeniu porcji próbki do chromatografu gazowego, rozdzieleniu węglowodorów C3-C4 od pozostałych składników, wykryciu ich przy pomocy detektora płomieniowo-jonizacyjnego (FID) i określeniu ich zawartości.
2.1. Określenie zawartości węglowodorów C3-C4 polega na dokonaniu identyfikacji składników mieszaniny na podstawie czasów retencji.
2.2. Sposób wykonywania badania, stosowane odczynniki i materiały, rodzaj aparatury, sposób pobierania i przygotowywania próbek, sposób przygotowywania aparatury, sposób wykonywania oznaczenia, obliczania i podawania wyników, precyzję oraz sposób sporządzania protokołu z badania określa norma PN-EN 27941 lub norma DIN 51619.
3. Zawartość 1,3-butadienu określa się metodą chromatografii gazowej, polegającą na wprowadzeniu porcji próbki do chromatografu gazowego, rozdzieleniu 1,3-butadienu od pozostałych składników, wykryciu go przy pomocy detektora płomieniowo-jonizacyjnego (FID) i określeniu jego zawartości.
3.1. Określenie zawartości 1,3-butadienu polega na dokonaniu identyfikacji składników mieszaniny na podstawie czasów retencji.
3.2. Sposób wykonywania badania, stosowane odczynniki i materiały, rodzaj aparatury, sposób pobierania i przygotowywania próbek, sposób przygotowywania aparatury, sposób wykonywania oznaczenia, obliczania i podawania wyników, precyzję oraz sposób sporządzania protokołu z badania określa norma DIN 51619.
VI. Procedurę postępowania w sprawach dotyczących precyzji metody badania oraz interpretacji wyników badań określa norma PN-EN ISO 4259-2.
Załącznik nr 7
SPOSÓB POBIERANIA PRÓBEK BIOKOMPONENTÓW
1. Próbki pobiera się ręcznie ze zbiornika lub z opakowania jednostkowego, gdy ich zawartość znajduje się w spoczynku, przy czym próbki bioetanolu (etanolu paliwowego) należy pobierać metodą ręczną zgodnie z normami PN-EN ISO 3170 lub PN-A-79527.
2. Próbki ze zbiornika należy pobierać w kolejności od lustra cieczy do jego dna, aby uniknąć zaburzeń w niższych poziomach cieczy.
3. Próbki z opakowania jednostkowego należy pobierać z wybranego poziomu cieczy lub jako próbkę reprezentatywną przekrojową.
4. Próbki pobiera się przy użyciu odpowiednich przyrządów do pobierania próbek.
4.1. Rodzaje przyrządów do pobierania próbek:
1) bioetanolu określa norma PN-A-79527 lub norma PN-EN ISO 3170;
2) estru metylowego określa norma PN-EN ISO 3170;
3) biowęglowodorów ciekłych określa norma PN-EN ISO 3170 lub PN-EN ISO 3171;
4) bio propanu-butanu określa norma PN-EN ISO 4257, która precyzuje procedurę pobierania próbek, warunki bezpieczeństwa w miejscu ich pobierania, związane z użytkowaniem pojemników, w których próbki są przechowywane, oraz podczas ich transportu, rodzaj aparatury oraz pojemników stosowanych do pobierania próbek, a także sposób przygotowania pojemników na próbki.
5. Próbki pobiera się do odpowiednich pojemników.
5.1. Pojemniki przeznaczone na próbki:
1) bioetanolu powinny być wykonane ze szkła, metalu lub z tworzyw sztucznych, chemicznie obojętnych w stosunku do bioetanolu (np. HTPE, PET);
2) estru metylowego powinny być wykonane ze stali nierdzewnej lub z tworzyw sztucznych, chemicznie obojętnych;
3) bio propanu-butanu powinny być wykonane ze stali nierdzewnej;
4) biowęglowodorów ciekłych powinny być metalowe lub szklane, wykonane z materiału niezawierającego ołowiu.
5.2. Pojemniki przeznaczone na próbki powinny:
1) mieć pojemność maksymalnie 5 dm³ w przypadku bioetanolu, estru metylowego oraz biowęglowodorów ciekłych oraz maksymalnie 7 dm³ w przypadku bio propanu-butanu;
2) być wyposażone w uszczelki lub mieć połączenia szczelne, zdolne do wytrzymania wewnętrznych ciśnień, powstających podczas normalnej ich eksploatacji;
3) mieć zamocowanie, umożliwiające ich zaplombowanie.
5.3. W przypadku pobierania próbek bio propanu-butanu – próbkę należy pobierać tylko z fazy ciekłej.
5.4. Pojemniki przeznaczone na próbki nie mogą być zabezpieczane przed korozją środkami wytworzonymi na bazie produktu naftowego.
5.5. Zamknięcie pojemników przeznaczonych na próbki ciekłych biokomponentów składa się z nakrętki z dopasowaną podkładką odporną na działanie pobieranego biokomponentu. Podkładki te nie mogą być wykonane z korka lub gumy.
6. Przyrządy do pobierania próbek oraz pojemniki przeznaczone na próbki powinny być wykonane z materiałów chemicznie obojętnych dla pobieranego biokomponentu.
7. Pojemnik przeznaczony na próbkę należy:
1) w przypadku bioetanolu, estru metylowego bądź węglowodorów ciekłych napełnić pojemniki tak, aby pozostało co najmniej 5% wolnej przestrzeni ze względu na rozszerzalność produktu. Pobrana próbka powinna być utrzymywana w stanie ciekłym, a stopień napełnienia próbnika powinien gwarantować jego bezpieczny transport i użytkowanie;
2) po napełnieniu biokomponentem natychmiast zamknąć, stosując zamknięcie zapewniające niezmienność parametrów jakościowych próbki.
8. Szczelność zamkniętego pojemnika przeznaczonego na próbkę ciekłego biokomponentu należy sprawdzić, odwracając go do góry dnem i trzymając w tej pozycji przez 30 sekund. Jeżeli zaobserwuje się wyciek ciekłego biokomponentu, należy wymienić zamknięcie pojemnika na nowe i ponownie sprawdzić jego szczelność. Sprawdzenie szczelności pojemnika ciśnieniowego na bio propan-butan należy przeprowadzić zgodnie z normą PN-EN ISO 4257.
8.1. W przypadku gdy wyciek biokomponentu nie ustaje, należy ponownie pobrać próbkę, używając nowego pojemnika wraz z nowym zamknięciem, i ponownie przeprowadzić ocenę szczelności pojemnika i zamknięcia.
9. Sposób postępowania przy pobieraniu próbek, postępowanie z próbkami oraz wymagania dotyczące bezpieczeństwa w tym zakresie określa: dla bioetanolu norma PN-EN ISO 3170 lub PN-A-79527, dla estru metylowego norma PN-EN ISO 3170, dla biowęglowodorów ciekłych norma PN-EN ISO 3170 lub PN-EN ISO 3171, dla bio propanu-butanu norma PN-EN ISO 4257.
- Data ogłoszenia: 2021-09-16
- Data wejścia w życie: 2021-10-01
- Data obowiązywania: 2021-10-01
- Dokument traci ważność: 2026-10-02

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA