REKLAMA
Dziennik Ustaw - rok 2020 poz. 931
ROZPORZĄDZENIE
MINISTRA ZDROWIA1)
z dnia 19 marca 2020 r.
w sprawie metod oznaczeń próbek niezbędnych do kontroli bezpieczeństwa produktów kosmetycznych2)
Na podstawie art. 25 ust. 3 ustawy z dnia 4 października 2018 r. o produktach kosmetycznych (Dz. U. poz. 2227) zarządza się, co następuje:
§ 1. [Metody oznaczeń próbek]
§ 2. [Wejście w życie]
Minister Zdrowia: wz. J. Szczurek-Żelazko
1) Minister Zdrowia kieruje działem administracji rządowej - zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 18 listopada 2019 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. poz. 2269).
2) Niniejsze rozporządzenie wdraża następujące dyrektywy:
1) pierwszą dyrektywę Komisji 80/1335/EWG z dnia 22 grudnia 1980 r. w sprawie zbliżania ustawodawstw Państw Członkowskich odnoszących się do metod analizy niezbędnych do kontrolowania składu produktów kosmetycznych (Dz. Urz. WE L 383 z 31.12.1980, str. 27; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 6, str. 109 oraz Dz. Urz. WE L 57 z 27.02.1987, str. 56; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 8, str. 268);
2) drugą dyrektywę Komisji 82/434/EWG z dnia 14 maja 1982 r. w sprawie zbliżenia ustawodawstw Państw Członkowskich odnoszących się do metod analizy niezbędnych do kontrolowania składu produktów kosmetycznych (Dz. Urz. WE L 185 z 30.06.1982, str. 1; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 6, str. 301 oraz Dz. Urz. WE L 108 z 28.04.1990, str. 92; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 10, str. 122);
3) trzecią dyrektywę Komisji 83/514/EWG z dnia 27 września 1983 r. w sprawie zbliżania ustawodawstw Państw Członkowskich dotyczących metod analizy niezbędnych do kontrolowania składu produktów kosmetycznych (Dz. Urz. WE L 291 z 24.10.1983, str. 9; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 7, str. 124);
4) czwartą dyrektywę Komisji 85/490/EWG z dnia 11 października 1985 r. w sprawie zbliżenia ustawodawstw Państw Członkowskich odnoszących się do metod analizy niezbędnych do kontrolowania składu produktów kosmetycznych (Dz. Urz. WE L 295 z 07.11.1985, str. 30; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 8, str. 46);
5) piątą dyrektywę Komisji 93/73/EWG z dnia 9 września 1993 r. w sprawie metod analizy niezbędnych do kontrolowania składu produktów kosmetycznych (Dz. Urz. WE L 231 z 14.09.1993, str. 34; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 12, str. 222);
6) szóstą dyrektywę Komisji 95/32/WE z dnia 7 lipca 1995 r. odnoszącą się do metod analizy niezbędnych do kontroli składu produktów kosmetycznych (Dz. Urz. WE L 178 z 28.07.1995, str. 20; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 15, str. 171);
7) siódmą dyrektywę Komisji 96/45/WE z dnia 2 lipca 1996 r. odnoszącą się do metod analizy niezbędnych do kontroli składu produktów kosmetycznych (Dz. Urz. WE L 213 z 22.08.1996, str. 8; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 17, str. 142).
3) Niniejsze rozporządzenie było poprzedzone rozporządzeniem Ministra Zdrowia z dnia 23 grudnia 2002 r. w sprawie określenia procedur pobierania próbek kosmetyków oraz procedur przeprowadzania badań laboratoryjnych (Dz. U. z 2003 r. poz. 107 oraz z 2004 r. poz. 2106), które utraciło moc z dniem 2 stycznia 2020 r. zgodnie z art. 63 ustawy z dnia 4 października 2018 r. o produktach kosmetycznych (Dz. U. poz. 2227).
Załącznik do rozporządzenia Ministra Zdrowia
z dnia 19 marca 2020 r. (poz. 931)
METODY OZNACZEŃ PRÓBEK NIEZBĘDNYCH DO KONTROLI BEZPIECZEŃSTWA PRODUKTÓW KOSMETYCZNYCH
I. POBIERANIE PRÓBEK PRODUKTÓW KOSMETYCZNYCH
1. PRZEDMIOT I ZAKRES ZASTOSOWANIA
Procedurę pobierania próbek produktów kosmetycznych opisano, mając na względzie ich analizę w różnych laboratoriach.
2. DEFINICJE
2.1. Próbka podstawowa
Część pobrana z partii przeznaczonej na sprzedaż.
2.2. Próbka ogólna
Suma wszystkich próbek podstawowych mających ten sam numer partii.
2.3. Próbka laboratoryjna
Reprezentatywna część próbki ogólnej przeznaczona do analizy w poszczególnych laboratoriach.
2.4. Próbka analityczna
Reprezentatywna część próbki laboratoryjnej wymagana do jednej analizy.
2.5. Pojemnik
Opakowanie, które zawiera produkt i jest z nim w stałym bezpośrednim kontakcie.
3. PROCEDURA POBIERANIA PRÓBEK
3.1. Próbki produktów kosmetycznych pobiera się w oryginalnych pojemnikach i przesyła się do laboratorium analitycznego bez otwierania.
3.2. Dla produktów kosmetycznych, które są dostarczane na rynek hurtowo lub sprzedawane detalicznie w pojemnikach różniących się od oryginalnego opakowania producenta, podaje się odpowiednie instrukcje pobierania próbek w miejscach ich stosowania lub sprzedaży.
3.3. Liczba próbek podstawowych, wymaganych do przygotowania próbki laboratoryjnej, ma być odpowiednia do metody analitycznej, jak i liczby analiz wykonywanych w każdym laboratorium.
4. IDENTYFIKACJA PRÓBKI
4.1. Próbki pieczętuje się w momencie pobierania i opisuje w sposób pozwalający na ich identyfikację.
4.2. Każda pobrana próbka podstawowa zawiera na etykiecie następujące informacje:
1) nazwę produktu kosmetycznego;
2) datę, godzinę i miejsce pobrania próbki;
3) nazwisko osoby odpowiedzialnej za pobranie próbki;
4) nazwę urzędu kontrolującego.
4.3. Z pobrania próbki sporządza się sprawozdanie.
5. PRZECHOWYWANIE PRÓBEK
5.1. Próbki podstawowe należy przechowywać zgodnie z zaleceniami producenta, jeżeli są podane na etykiecie.
5.2. Jeżeli nie wymieniono innych warunków, próbki laboratoryjne należy przechowywać w ciemności, w temperaturze między 10–25°C.
5.3. Próbek podstawowych nie wolno otwierać przed rozpoczęciem analizy.
II. PRAKTYCZNE PRZYGOTOWANIE PRÓBEK LABORATORYJNYCH
1. OGÓLNE ZASADY
1.1. W każdym przypadku gdy to możliwe, analizę wykonuje się na każdej próbce podstawowej. Jeżeli próbka podstawowa jest zbyt mała, należy posługiwać się minimalną liczbą próbek podstawowych. Przed pobraniem próbki laboratoryjnej próbki podstawowe powinny być ze sobą starannie wymieszane.
1.2. Otworzyć pojemnik pod osłoną gazu obojętnego, jeżeli wymaga tego metoda analityczna, i możliwie szybko pobrać odpowiednią liczbę próbek laboratoryjnych oraz niezwłocznie przeprowadzić ich analizę. Jeżeli próbka podstawowa ma być zachowana, pojemnik należy szczelnie zamknąć pod osłoną gazu obojętnego.
1.3. Produkty kosmetyczne mogą być wytwarzane w postaci ciekłej lub stałej, a także półstałej. Jeżeli nastąpiła separacja faz pierwotnie homogenicznego produktu, przed pobraniem próbki laboratoryjnej należy go ponownie ujednorodnić.
1.4. Jeżeli produkty kosmetyczne trafiają do sprzedaży specjalną drogą, czego konsekwencją jest brak możliwości zastosowania się do niniejszej instrukcji, i jeżeli nie ma uregulowań dotyczących odpowiednich metod badawczych, możliwe jest użycie oryginalnej procedury pod warunkiem, że zostanie ona sformułowana na piśmie, a opis ten będzie stanowił część raportu analitycznego.
2. CIECZE
2.1. Produkty te mogą występować w takich formach, jak roztwory w oleju, w alkoholu i w wodzie, wody toaletowe, płyny lub mleczka. Mogą być one pakowane w butelki, ampułki lub tuby.
2.2. Pobieranie próbki laboratoryjnej:
1) przed otwarciem pojemnik energicznie wstrząsnąć;
2) otworzyć pojemnik;
3) wlać kilka mililitrów cieczy do probówki w celu obejrzenia, czy materiał nadaje się do pobrania próbki laboratoryjnej;
4) ponownie zamknąć pojemnik lub
5) pobrać wymagane próbki laboratoryjne;
6) starannie ponownie zamknąć pojemnik.
3. SUBSTANCJE PÓŁSTAŁE
3.1. Produkty mogą występować w takich formach, jak pasty, kremy, sztywne emulsje i żele. Mogą być pakowane w tuby, butelki z tworzyw sztucznych lub słoiki.
3.2. Pobieranie próbki laboratoryjnej może następować w różny sposób:
3.2.1. W przypadku pojemników z wąską szyjką: usunąć przynajmniej pierwszy centymetr produktu oraz wycisnąć próbkę laboratoryjną i niezwłocznie zamknąć pojemnik.
3.2.2. W przypadku pojemników z szeroką szyjką: zdjąć równomiernie całą wierzchnią warstwę produktu i usunąć ten materiał oraz pobrać próbkę laboratoryjną i niezwłocznie ponownie zamknąć pojemnik.
4. CIAŁA STAŁE
4.1. Produkty te mogą występować w takich formach, jak sypkie proszki, sprasowane proszki, sztyfty i mogą być pakowane w pojemniki różnego rodzaju.
4.2. Pobieranie próbki laboratoryjnej może następować w różny sposób:
4.2.1. W przypadku sypkiego proszku: wstrząsnąć energicznie przed odkorkowaniem lub otwarciem. Otworzyć i pobrać próbkę laboratoryjną.
4.2.2. W przypadku prasowanego proszku lub sztyftu: usunąć wierzchnią warstwę przez równomierne drapanie. Pobrać próbkę laboratoryjną spod spodu. 5. PRODUKTY W POJEMNIKACH POD CIŚNIENIEM (dozowniki aerozolowe)
5.1. Produkty te są zdefiniowane w przepisach wydanych na podstawie art. 10 ustawy z dnia 30 sierpnia 2002 r. o systemie oceny zgodności (Dz. U. z 2019 r. poz. 155) dotyczących szczegółowych wymagań dla wyrobów aerozolowych.
5.2. Próbka laboratoryjna
Po energicznym wstrząśnięciu reprezentatywną część zawartości dozownika aerozolowego przenieść za pomocą odpowiedniego łącznika (patrz: przykładowy rys. 1; w szczególnych przypadkach metoda analityczna może wymagać użycia innych łączników) do szklanej butelki (rys. 4) pokrytej tworzywem sztucznym, wyposażonej w zawór aerozolowy nieposiadający rurki zanurzeniowej. Podczas przenoszenia próbki butelkę trzyma się zaworem skierowanym w dół. Takie przeniesienie próbki zapewnia dobrą widoczność zawartości pojemnika i odpowiada jednemu z następujących czterech przypadków:
5.2.1. Wyrób aerozolowy w postaci jednorodnego roztworu do bezpośredniej analizy.
5.2.2. Wyrób aerozolowy składający się z dwóch faz ciekłych. Każda z faz może być analizowana po oddzieleniu dolnej fazy do drugiej pomocniczej butelki do przenoszenia. W tym przypadku pierwsza butelka do przenoszenia jest skierowana zaworem w dół. W takim przypadku dolna faza jest często fazą wodną i nie zawiera propelenta (np. wyrób z butanem i wodą).
5.2.3. Wyrób aerozolowy zawierający proszek w zawiesinie. Fazę ciekłą można analizować po oddzieleniu proszku.
5.2.4. Wyrób w postaci piany lub kremu. Najpierw należy odważyć do pomocniczej butelki do przenoszenia od 5 do 10 g 2-metoksyetanolu. Substancja ta zabezpiecza przed tworzeniem się piany podczas operacji odgazowania i wtedy można usuwać propelent bez strat cieczy.
5.3. Wyposażenie
Łącznik (rys. 1) jest wykonany z duraluminium lub mosiądzu. Jest przeznaczony do połączenia różnych zaworów przez łącznik polietylenowy (patrz: przykładowe rys. 2 i 3). Można używać innych łączników.
Pomocnicza butelka do przenoszenia (rys. 4) jest wykonana z białego szkła pokrytego zewnętrzną warstwą ochronną z przezroczystego tworzywa. Jej pojemność wynosi od 50 do 100 ml i jest ona wyposażona w zawór aerozolowy bez rurki zanurzeniowej.
5.4. Metoda
Aby można było przenieść wystarczającą ilość próbki, z butelki do przenoszenia musi być usunięte powietrze. W tym celu należy wprowadzić przez łącznik ok. 10 ml dichlorodifluorometanu lub butanu (zależnie od rodzaju badanego wyrobu aerozolowego) i całkowicie odgazować aż do zaniku fazy ciekłej, trzymając butelkę do przenoszenia zaworem skierowanym do góry. Usunąć łącznik. Zważyć butelkę do przenoszenia („a” gramów). Energicznie wytrząsnąć pojemnik aerozolowy, z którego ma być pobierana próbka. Połączyć łącznik z zaworem na pojemniku aerozolowym z badaną zawartością (zawór skierowany do góry), dopasować butelkę do przenoszenia próbki (szyjką skierowaną w dół) do łącznika i nacisnąć. Napełnić butelkę do przenoszenia do ok. 2/3 pojemności. Jeżeli przenoszenie przedwcześnie ustaje na skutek wyrównania ciśnień, można je wznowić przez chłodzenie butelki do przenoszenia. Usunąć łącznik, zważyć napełnioną butelkę („b” gramów) i oznaczyć masę przeniesionej próbki m1 (m1 = b – a). Otrzymaną w ten sposób próbkę można użyć do:
1) normalnej analizy chemicznej;
2) analizy składników lotnych metodą chromatografii gazowej.
5.4.1. Analiza chemiczna
Trzymając butelkę do przenoszenia zaworem do góry, należy postępować następująco:
1) odgazować; jeżeli czynność ta powoduje powstawanie piany, należy użyć butelki do przenoszenia, do której uprzednio za pomocą strzykawki dodano przez łącznik dokładnie odważoną ilość (od 5 do 10 g) 2-metoksyetanolu;
2) bez straty próbki zakończyć usuwanie lotnych składników przez wytrząsanie w łaźni wodnej o temperaturze 40°C, zdjąć łącznik;
3) ponownie zważyć butelkę do przenoszenia („c” gramów) w celu ustalenia ciężaru pozostałości m2 (m2 = c – a);
(Uwaga: Przy obliczaniu ciężaru pozostałości należy odjąć ciężar ewentualnie użytego 2-metoksyetanolu);
4) otworzyć butelkę do przenoszenia przez zdjęcie zaworu;
5) całkowicie rozpuścić pozostałość w znanej ilości odpowiedniego rozpuszczalnika;
6) przeprowadzić wymagane oznaczenie na części pobranej próbki.
Wzory do obliczeń są następujące:
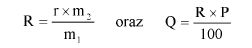
gdzie:
m1 = masa aerozolu pobrana do butelki do przenoszenia,
m2 = masa pozostałości po ogrzaniu do temperatury 40°C,
r = procent określonej substancji w m2 (ustalony według właściwej metody),
R = procent określonej substancji w aerozolu, jaki otrzymano do analizy,
Q = całkowita masa określonej substancji w dozowniku aerozolowym,
P = początkowa masa netto dozownika aerozolowego (próbka podstawowa).
5.4.2. Analiza lotnych składników metodą chromatografii gazowej.
5.4.2.1. Zasady
Za pomocą strzykawki do chromatografii gazowej pobrać właściwą ilość materiału z butelki do przenoszenia. Następnie wstrzyknąć zawartość strzykawki do chromatografu gazowego.
5.4.2.2. Oprzyrządowanie
Precyzyjna strzykawka o pojemności 25 µl lub 50 µl szeregu A2 (rys. 5) do pobierania próbek dla chromatografii gazowej lub równoważna. Ta strzykawka jest wyposażona w zawór odcinający przy nasadzie igły. Strzykawka jest połączona z butelką do przenoszenia za pomocą łącznika przy butelce i polietylenowej rurki (długość 8 mm, wewnętrzna średnica 2,5 mm) przy strzykawce.
5.4.2.3. Metoda
Po pobraniu do butelki do przenoszenia właściwej ilości produktu aerozolowego dopasować stożkowe zakończenie strzykawki do butelki, jak opisano w pkt 5.4.2.2. Otworzyć zawór i nabrać właściwą ilość cieczy. Usunąć pęcherzyki gazu przez kilkakrotne poruszenie tłokiem (schłodzić strzykawkę, jeżeli to konieczne). Jeżeli strzykawka zawiera właściwą ilość pozbawionej pęcherzyków cieczy, zamknąć zawór i odłączyć strzykawkę od butelki do przenoszenia. Założyć igłę, włożyć strzykawkę do iniektora chromatografu gazowego i wstrzyknąć.
5.4.2.4. Wzorzec wewnętrzny
Jeżeli potrzebny jest wzorzec wewnętrzny, to jest on wprowadzany do butelki do przenoszenia (za pomocą zwykłej szklanej strzykawki z użyciem łącznika).
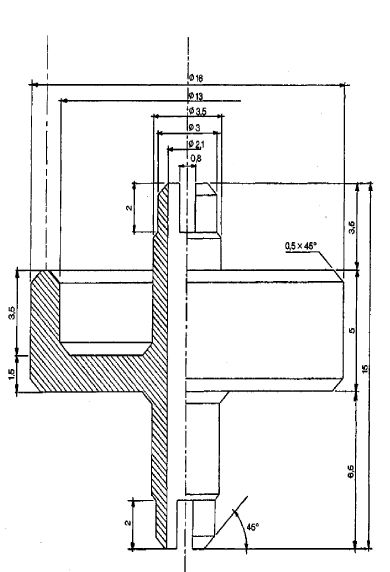
Rys. 1 Łącznik P1
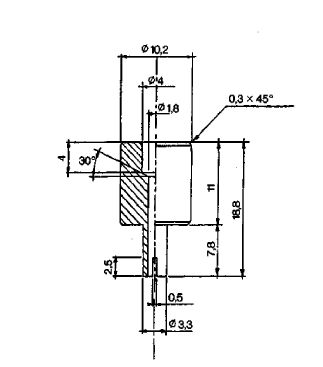
Rys. 2
Łącznik Μ2 do przenoszenia między zaworem męskim i żeńskim
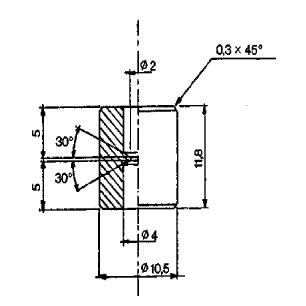
Rys. 3
Łącznik M1 do przenoszenia między dwoma zaworami żeńskimi
Rys. 4
Butelka do przenoszenia
Pojemność od 50 do 100 ml
Rys. 5
Strzykawka do gazu pod ciśnieniem
III. IDENTYFIKOWANIE I OZNACZANIE ZAWARTOŚCI WOLNYCH WODOROTLENKÓW SODU I POTASU
1. CEL I ZAKRES METODY
Metoda podaje dokładną procedurę identyfikowania produktów kosmetycznych zawierających znaczące ilości wolnych wodorotlenków sodu (INCI: sodium hydroxide) i potasu (INCI: potassium hydroxide) i oznaczenia takich wolnych wodorotlenków sodu i/lub potasu w środkach do prostowania włosów i w rozpuszczalnikowych środkach do usuwania skórek paznokci.
2. DEFINICJA
Wolny wodorotlenek sodu i wolny wodorotlenek potasu są określane poprzez objętość (ilość) standardowego roztworu kwasu, wymaganą do zobojętnienia produktu w podanych warunkach, a otrzymaną ilość wyraża się jako % m/m wolnego wodorotlenku sodu.
3. ZASADA
Próbka zostaje rozpuszczona lub zdyspergowana w wodzie i miareczkowana standardowym roztworem kwasu. Wartość pH jest rejestrowana równocześnie z dodawaniem kwasu do roztworu wodorotlenku sodu lub potasu. W przypadku prostych roztworów wodorotlenków sodu lub potasu punktem końcowym jest wyraźny wzrost szybkości zmian wartości pH. Krzywa miareczkowania może być niewyraźna w obecności:
1) amoniaku lub innych słabych zasad organicznych, które same mają raczej spłaszczoną krzywą miareczkowania (w tej metodzie amoniak usuwa się przez odparowanie pod zmniejszonym ciśnieniem, ale w pokojowej temperaturze);
2) soli słabych kwasów, które mogą powodować wzrost krzywej miareczkowania z kilkoma punktami przegięcia (w tych przypadkach tylko pierwsza część krzywej do pierwszego z tych punktów przegięcia odpowiada zobojętnianiu jonu wodorotlenowego pochodzącego z wolnego wodorotlenku sodu lub potasu).
Podano alternatywną procedurę miareczkowania, odpowiednią dla przypadku, gdy zauważa się nadmierne oddziaływanie soli słabych kwasów nieorganicznych. Chociaż istnieje teoretyczna możliwość, że inne rozpuszczalne mocne zasady, np. wodorotlenek litu, czwartorzędowy wodorotlenek amonowy, mogłyby być obecne, dając wzrost pH do wysokich wartości, jednak obecność ich w takim rodzaju wyrobów jak produkty kosmetyczne jest wysoce nieprawdopodobna.
4. IDENTYFIKOWANIE
4.1. Odczynniki
Standardowy alkaliczny roztwór buforowy o pH = 9,18 w temperaturze 25°C: 0,05 M roztwór tetraboranu disodu.
4.2. Aparatura
4.2.1. Zwykłe szklane wyposażenie laboratoryjne.
4.2.2. Pehametr.
4.2.3. Szklana elektroda membranowa.
4.2.4. Standardowa kalomelowa elektroda odniesienia.
4.3. Procedura
Skalibrować pehametr za pomocą elektrod z zastosowaniem standardowego roztworu buforowego. Przygotować 10% roztwór lub dyspersję analizowanego wyrobu w wodzie i przesączyć go. Zmierzyć pH. Jeżeli pH wynosi 12 lub więcej, należy wykonać oznaczanie zawartości ilościowej.
5. OZNACZANIE
5.1. Miareczkowanie w środowisku wodnym.
5.1.1. Odczynnik
5.1.1.1. Standardowy 0,1 N kwas chlorowodorowy.
5.1.2. Aparatura
5.1.2.1. Zwykłe szklane wyposażenie laboratoryjne.
5.1.2.2. Pehametr, korzystnie, jeżeli wyposażony w rejestrator.
5.1.2.3. Szklana elektroda membranowa.
5.1.2.4. Standardowa kalomelowa elektroda odniesienia.
5.1.3. Procedura
Zważyć dokładnie w zlewce o pojemności 150 ml próbkę wielkości od 0,5 do 1,0 g. Jeżeli w próbce znajduje się amoniak, dodać kilka ziaren porowatych, umieścić zlewkę w eksykatorze próżniowym, usuwać amoniak za pomocą pompy wodnej tak długo, aż jego zapach będzie niewyczuwalny (ok. trzech godzin). Dodać 100 ml wody, rozpuścić lub zdyspergować pozostałość i miareczkować 0,1 N roztworem kwasu chlorowodorowego (5.1.1.1), rejestrując zmiany pH (5.1.2.2).
5.1.4. Obliczenie
Umiejscowić punkty przegięcia na krzywych miareczkowania. Jeżeli pierwszy punkt przegięcia występuje przy pH niższym niż 7, próbka nie zawiera wodorotlenku sodu lub potasu.
Jeżeli na krzywej znajdują się dwa punkty lub więcej punktów przegięcia, tylko pierwszy punkt ma znaczenie. Zanotować objętość roztworu zużytego do miareczkowania do pierwszego punktu przegięcia.
Oznaczyć symbolem V objętość roztworu zużytego do miareczkowania, w mililitrach.
Oznaczyć symbolem M masę próbki analitycznej, w gramach.
Zawartość wodorotlenku sodu i/lub potasu w próbce wyrażoną jako % m/m wodorotlenku sodu oblicza się, stosując wzór: % = 0,4 (v/m).
Może powstać sytuacja, w której pomimo oznak obecności znaczącej ilości wodorotlenków sodu i/lub potasu krzywa miareczkowania nie wykazuje wyraźnego punktu przegięcia. W takim przypadku należy powtórzyć oznaczenie w izopropanolu.
5.2. Miareczkowanie w izopropanolu.
5.2.1. Odczynniki
5.2.1.1. Izopropanol.
5.2.1.2. Standardowy 1,0 N wodny roztwór kwasu chlorowodorowego.
5.2.1.3. 0,1 N roztwór kwasu chlorowodorowego w izopropanolu przygotowany bezpośrednio przed użyciem przez rozcieńczenie 1,0 N wodnego roztworu kwasu chlorowodorowego izopropanolem.
5.2.2. Aparatura
5.2.2.1. Zwykłe szklane wyposażenie laboratoryjne.
5.2.2.2. Pehametr, korzystnie, jeżeli z rejestratorem.
5.2.2.3. Szklana elektroda membranowa.
5.2.2.4. Standardowa kalomelowa elektroda odniesienia.
5.2.3. Procedura
Zważyć dokładnie w zlewce o pojemności 150 ml próbkę wielkości od 0,5 do 1,0 g. Jeżeli w próbce znajduje się amoniak, dodać kilka ziaren porowatych, umieścić zlewkę w eksykatorze próżniowym, usuwać amoniak za pomocą pompy wodnej tak długo, aż jego zapach będzie niewyczuwalny (ok. trzech godzin).
Dodać 100 ml izopropanolu, rozpuścić lub zdyspergować pozostałość i miareczkować 0,1 N kwasem chlorowodorowym (5.2.1.3), rejestrując odczytane zmiany pH (5.2.2.2).
5.2.4. Obliczenie
Jak opisano w pkt 5.1.4.
Pierwszy punkt przegięcia występuje przy odczytanej wartości pH ok. 9.
5.3. POWTARZALNOŚĆ (patrz: norma ISO/DIS 5725)
Dla wodorotlenku sodu lub potasu na poziomie 5% (m/m), wyrażonego jako wodorotlenek sodu, różnica między wynikami dwóch oznaczeń, wykonywanych równolegle na tej samej próbce, nie powinna przekraczać wartości bezwzględnej 0,25%.
IV. IDENTYFIKOWANIE I OZNACZANIE ZAWARTOŚCI KWASU SZCZAWIOWEGO I JEGO SOLI ALKALICZNYCH W PRODUKTACH KOSMETYCZNYCH DO PIELĘGNACJI WŁOSÓW
1. CEL I ZAKRES METODY
Metoda jest odpowiednia do identyfikowania i oznaczania kwasu szczawiowego (INCI: oxalic acid) i jego soli alkalicznych w produktach kosmetycznych do pielęgnacji włosów. Może być stosowana dla bezbarwnych wodnych/alkoholowych roztworów i płynów, które zawierają ok. 5% kwasu szczawiowego lub równoważną ilość alkalicznych szczawianów.
2. DEFINICJA
Zawartość kwasu szczawiowego lub jego soli alkalicznych oznaczona według tej metody jest wyrażona jako zawartość w procentach masowych (m/m) wolnego kwasu szczawiowego w próbce.
3. ZASADA
Po usunięciu wszystkich obecnych anionowych środków powierzchniowo czynnych chlorowodorkiem p-toluidyny kwas szczawiowy i/lub szczawiany są wytrącane jako szczawian wapnia, po czym roztwór jest przesączany. Osad jest rozpuszczany w kwasie siarkowym i miareczkowany roztworem manganianu (VII) potasu.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. 5% (m/m) roztwór octanu amonu.
4.2. 10% (m/m) roztwór chlorku wapnia.
4.3 95% (v/v) etanol.
4.4. Tetrachlorek węgla.
4.5. Eter dietylowy.
4.6. 6,8% (m/m) roztwór dichlorowodorku p-toluidyny.
4.7. 0,1 N roztwór manganianu (VII) potasu.
4.8. 20% (m/m) kwas siarkowy (VI).
4.9. 10% (m/m) kwas chlorowodorowy.
4.10. Trójwodny octan sodu (trihydrat octanu sodu).
4.11. Kwas octowy lodowaty.
4.12. Kwas siarkowy (VI) (1:1).
4.13. Nasycony roztwór wodorotlenku baru.
5. APARATURA
5.1. Rozdzielacze, 500 ml.
5.2. Zlewki, 50 ml i 600 ml.
5.3. Tygle z filtrem szklanym G-4.
5.4. Cylindry miarowe, 25 ml i 100 ml.
5.5. Pipety, 10 ml.
5.6. Kolby ssawkowe, 500 ml.
5.7. Próżniowa pompa wodna.
5.8. Termometr o zakresie skali od 0 do 100°C.
5.9. Mieszadło magnetyczne z elementem grzewczym.
5.10. Pręty magnetyczne pokryte teflonem.
5.11. Biureta, 25 ml.
5.12. Kolby stożkowe, 250 ml.
6. PROCEDURA
6.1. Odważyć 6 do 7 g próbki do zlewki o pojemności 50 ml, doprowadzić do pH 3 rozcieńczonym kwasem chlorowodorowym (4.9) i przemyć w rozdzielaczu 100 ml wody destylowanej. Dodawać stopniowo 25 ml etanolu (4.3), 25 ml roztworu dichlorowodorku p-toluidyny (4.6) i od 25 do 30 ml czterochlorku węgla (4.4) i wytrząsnąć energicznie mieszaninę.
6.2. Po rozdzieleniu faz usunąć dolną (organiczną) fazę, powtórzyć ekstrakcję, używając reagentów podanych w pkt 6.1, i znowu usunąć fazę organiczną.
6.3. Przelać wodny roztwór do zlewki o pojemności 600 ml i usunąć jeszcze obecny czterochlorek węgla przez ogrzewanie roztworu do wrzenia.
6.4. Dodać 50 ml roztworu octanu amonu (4.1), doprowadzić roztwór do wrzenia (5.9) i wmieszać 10 ml gorącego roztworu chlorku wapnia (4.2) do wrzącego roztworu; pozwolić na wytrącenie się osadu.
6.5. Sprawdzić, czy wytrącenie osadu jest całkowite, dodać kilka kropli roztworu chlorku wapnia (4.2), pozostawić do schłodzenia do temperatury pokojowej i następnie wmieszać 200 ml etanolu (4.3); (5.10) pozostawić do odstania na 30 minut.
6.6. Przesączyć ciecz przez tygiel z filtrem szklanym (5.3), przenieść osad z małą ilością gorącej wody (od 50 do 60°C) do tygla z filtrem szklanym i przemyć osad zimną wodą.
6.7. Przemyć osad pięć razy małą ilością etanolu (4.3) i potem pięć razy małą ilością eteru etylowego (4.5) i rozpuścić osad w 50 ml gorącego kwasu siarkowego (VI) (4.8) przez przeciąganie tego ostatniego przez szklany filtr tygla pod zmniejszonym ciśnieniem.
6.8. Przenieść roztwór bez strat do kolby stożkowej (5.12) i miareczkować roztworem manganianu (VII) potasu (4.7) aż do wystąpienia jasnoróżowego zabarwienia.
7. OBLICZENIE
Zawartość kwasu szczawiowego w próbce wyrażoną jako procent masowy oblicza się według wzoru:
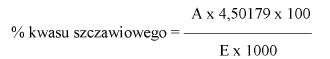
gdzie:
A – objętość 0,1 N roztworu manganianu (VII) potasu zużyta do miareczkowania (6.8),
E – masa próbki analitycznej w gramach (6.1),
4,50179 – współczynnik przeliczeniowy kwasu szczawiowego.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości kwasu szczawiowego wynoszącej ok. 5% różnica między wynikami z dwóch równolegle prowadzonych analiz na tej samej próbce nie powinna przekraczać wartości bezwzględnej 0,15%.
9. IDENTYFIKOWANIE
9.1. Zasada
Kwas szczawiowy i/lub szczawiany wytrącane są w postaci szczawianu wapnia i rozpuszczane w kwasie siarkowym (VI). Do roztworu dodaje się niewielką ilość roztworu manganianu (VII) potasu, który się odbarwia i powoduje tworzenie się dwutlenku węgla. Kiedy powstały dwutlenek węgla przechodzi przez roztwór wodorotlenku baru, tworzy się biały osad (zmętnienie) węglanu baru.
9.2. Procedura
9.2.1. Poddać próbkę przeznaczoną do analizy działaniu opisanemu w pkt 6.1–6.3, które usunie każdy obecny środek powierzchniowo czynny.
9.2.2. Dodać nieco, na czubku łopatki, octanu sodu (4.10) do ok. 10 ml roztworu otrzymanego zgodnie z pkt 9.2.1 i zakwasić roztwór kilkoma kroplami kwasu octowego lodowatego (4.11).
9.2.3. Dodać 10% roztwór chlorku wapnia (4.2) i odsączyć. Rozpuścić osad szczawianu wapnia w 2 ml kwasu siarkowego (1:1) (4.12).
9.2.4. Przenieść roztwór do probówki i dodać kroplami ok. 0,5 ml 0,1 N roztworu manganianu (VII) potasu (4.7). Jeżeli szczawian jest obecny, roztwór traci kolor, najpierw stopniowo, później gwałtownie.
9.2.5. Bezpośrednio po dodaniu manganianu (VII) potasu umieścić odpowiednią szklaną rurkę z korkiem nad probówką, ogrzewać lekko zawartość i zbierać powstający dwutlenek węgla w nasyconym roztworze wodorotlenku baru (4.13). Pojawienie się po 3–5 minutach mlecznego zmętnienia węglanu baru świadczy o obecności kwasu szczawiowego.
V. OZNACZANIE ZAWARTOŚCI CHLOROFORMU W PAŚCIE DO ZĘBÓW
1. CEL I ZAKRES METODY
Metoda jest stosowana do oznaczania chloroformu w paście do zębów za pomocą chromatografii gazowej. Metoda jest odpowiednia do oznaczania chloroformu na poziomie 5% lub poniżej.
2. DEFINICJA
Zawartość chloroformu oznaczona tą metodą jest wyrażona jako procent masowy w wyrobie.
3. ZASADA
Otrzymuje się zawiesinę pasty do zębów w mieszaninie dimetyloformamidu/metanolu, do której dodaje się znaną ilość acetonitrylu jako standard wewnętrzny. Po odwirowaniu próby faza ciekła jest analizowana metodą chromatografii gazowej i obliczana jest zawartość chloroformu.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. Porapak Q, Chromosorb 101 lub równoważny, 80 do 100 mesh.
4.2. Acetonitryl.
4.3. Chloroform.
4.4. Dimetyloformamid.
4.5. Metanol.
4.6. Roztwór standardu wewnętrznego
Do 50 ml kolbki miarowej dodać pipetą 5 ml dimetyloformamidu (4.4) i dodać ok. 300 mg (M mg) acetonitrylu, dokładnie odważonego. Uzupełnić do kreski dimetyloformamidem i wymieszać.
4.7. Roztwór do oznaczania względnego współczynnika odpowiedzi. Do 10 ml kolbki miarowej dodać pipetą dokładnie 5 ml roztworu wewnętrznego standardu (4.6) i dodać ok. 300 mg (M1 mg) chloroformu, dokładnie odważonego. Uzupełnić do kreski dimetyloformamidem i wymieszać.
5. APARATURA I WYPOSAŻENIE
5.1. Waga analityczna.
5.2. Chromatograf gazowy z detektorem płomieniowo-jonizacyjnym.
5.3. Strzykawka o pojemności 5 do 10 µl i kalibracją co 0,1 µl.
5.4. Pipety ze zbiornikiem gruszkowym o pojemności 1, 4 i 5 ml.
5.5. Kolby miarowe 10 i 50 ml.
5.6. Probówki, ok. 20 ml, z nakrętką, Sovirel France nr 20 lub równoważne. Nakrętka ma wewnętrzną płytkę uszczelniającą, pokrytą z jednej strony teflonem.
5.7. Wirówka.
6. PROCEDURA
6.1. Warunki chromatografii gazowej.
6.1.1. Kolumna szklana długości 150 cm, średnicy wewnętrznej 4 mm, średnicy zewnętrznej 6 mm.
6.1.2. Wypełnić kolumnę wypełnieniem Porapak Q, Chromosorb 101 lub równoważnym 80 do 100 mesh (4.1) za pomocą wibratora.
6.1.3. Detektor płomieniowo-jonizacyjny: należy nastawić jego czułość tak, aby przy wprowadzeniu 3 µl roztworu według pkt 4.7 wysokość piku acetonitrylu wynosiła ok. trzech czwartych pełnego zakresu wysokości piku.
6.1.4. Gazy:
Nośnik: azot, szybkość przepływu 65 ml/min.
Pomocniczy: nastawić przepływ gazów do detektora tak, aby przepływ powietrza lub tlenu był pięć do dziesięciu razy większy niż wodoru.
6.1.5. Temperatura:
1) dozownik: 210°C;
2) detektor: 210°C;
3) piec kolumny: 175°C.
6.1.6. Szybkość przesuwu papieru ok. 100 cm na godzinę.
6.2. Przygotowanie próbki
Pobrać próbkę do analizy z nieotwieranej jeszcze tubki. Usunąć najpierw jedną trzecią zawartości, zamknąć tubę, wymieszać starannie pastę i następnie pobrać próbkę analityczną.
6.3. Oznaczenie
6.3.1. Do probówki z nakrętką (5.6) odważyć 6 do 7 g (M0 g) z dokładnością do 10 mg pasty do zębów, przygotowanej zgodnie z pkt 6.2, i dodać trzy małe szklane ziarenka.
6.3.2. Do probówki dodać pipetą dokładnie 5 ml roztworu wewnętrznego standardu (4.6), 4 ml dimetyloformamidu (4.4) i 1 ml metanolu (4.5), zamknąć probówkę i wymieszać.
6.3.3. Wytrząsnąć zamkniętą probówkę w mechanicznej wytrząsarce w ciągu pół godziny i odwirować ją w ciągu 15 minut z taką szybkością, aby otrzymać wyraźny rozdział faz.
Uwaga: Czasami zdarza się, że faza ciekła jest mętna po wirowaniu. Pewną poprawę można uzyskać przez dodanie 1 do 2 g chlorku sodu do fazy ciekłej, pozostawienie do odstania i ponowne odwirowanie.
6.3.4. Wprowadzić do chromatografu 3 µl roztworu opisanego w pkt 6.3.3 w warunkach jak w pkt 6.1. Powtórzyć tę operację w warunkach opisanych powyżej, można podać następujący czas retencji jako wartość orientacyjną:
1) metanol – średnio 1 minuta;
2) acetonitryl – średnio 2,5 minuty;
3) chloroform – średnio 6 minut;
4) dimetyloformamid > 15 minut.
6.3.5. Oznaczenie względnego współczynnika odpowiedzi
Wprowadzić 3 µl roztworu opisanego w pkt 4.7 dla oznaczenia tego współczynnika.
Powtórzyć operację. Oznaczać względny współczynnik odpowiedzi codziennie.
7. OBLICZENIA
7.1. Obliczenie względnej odpowiedzi.
7.1.1. Zmierzyć wysokość i szerokość w połowie wysokości pików acetonitrylu i chloroformu i obliczyć powierzchnie obu pików, zgodnie z wzorem: wysokość x szerokość w połowie wysokości.
7.1.2. Oznaczyć powierzchnie pików acetonitrylu i chloroformu w chromatogramie, otrzymanym zgodnie z pkt 6.3.5 i obliczyć względną odpowiedź fs za pomocą następującego wzoru:
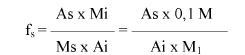
gdzie:
fs = współczynnik względnej odpowiedzi dla chloroformu,
As = powierzchnia piku chloroformu (6.3.5),
Ai = powierzchnia piku acetonitrylu (6.3.5),
Ms = ilość chloroformu w mg na 10 ml roztworu, jak w pkt 6.3.5 (= M1),
Mi = ilość acetonitrylu w mg na 10 ml roztworu, jak w pkt 6.3.5 (= 0,1 M).
Obliczyć średnią z otrzymanych wyników.
7.2. Obliczenie zawartości chloroformu.
7.2.1. Obliczyć zgodnie z pkt 7.1.1 powierzchnie pików chloroformu i acetonitrylu na chromatogramie otrzymanym w procedurze opisanej w pkt 6.3.4.
7.2.2. Obliczyć zawartość chloroformu w paście do zębów za pomocą następującego wzoru:
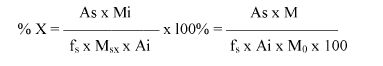
gdzie:
% X = zawartość chloroformu w paście do zębów wyrażona jako procent wagowy,
As = powierzchnia piku chloroformu (6.3.4),
Ai = powierzchnia piku acetonitrylu (6.3.4),
Msx = masa w mg próbki omówionej w pkt 6.3.1 (= 1000 M0),
Mi = ilość acetonitrylu w mg na 10 ml roztworu otrzymanego zgodnie z pkt 6.3.2 (= 0,1 M).
Obliczyć średnią oznaczoną zawartość i przedstawić wynik z dokładnością do 0,1%.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości chloroformu wynoszącej ok. 3% różnica między wynikami dwóch równoległych oznaczeń, wykonanych dla tej samej próbki, nie powinna przekroczyć bezwzględnej wartości 0,3%.
VI. OZNACZANIE CYNKU
1. CEL I ZAKRES METODY
Metoda jest odpowiednia do oznaczania cynku w produktach kosmetycznych, występującego jako chlorek, siarczan lub 4-hydroksybenzenosulfonian cynku lub jako połączenie kilku z tych soli cynku.
2. DEFINICJA
Zawartość cynku w próbce jest oznaczana grawimetrycznie jako bis-(2-metylochinolin-8-olano)-cynk i wyrażana jako procent wagowy cynku w próbce.
3. ZASADA
Cynk znajdujący się w roztworze jest w środowisku kwaśnym wytrącony jako bis-(2-metylochinolin-8-olano)-cynk. Po odsączeniu osad suszy się i waży.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. 25% (m/m) stężony amoniak, d 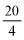
4.2. Lodowaty kwas octowy.
4.3. Octan amonu.
4.4. 2-metylochinolin-8-ol.
4.5. 6% (m/v) roztwór amoniaku
Przenieść 240 g stężonego amoniaku (4.1) do 1000 ml kolby miarowej, uzupełnić wodą destylowaną do kreski i wymieszać.
4.6. 0,2 M roztwór octanu amonu
Rozpuścić 15,4 g octanu amonu (4.3) w wodzie destylowanej w kolbie miarowej 1000 ml, uzupełnić wodą do kreski i wymieszać.
4.7. Roztwór 2-metylochinolin-8-olu
Rozpuścić 5 g 2-metylochinolin-8-olu w 12 ml kwasu octowego lodowatego i następnie przenieść z destylowaną wodą do 1000 ml kolby. Rozcieńczyć wodą do kreski i wymieszać.
5. APARATURA i WYPOSAŻENIE
5.1. Kolby miarowe, 100 i 1000 ml.
5.2. Zlewki, 400 ml.
5.3. Cylindry miarowe, 50 i 150 ml.
5.4. Pipety kalibrowane, 10 ml.
5.5. Tygle z filtrem G-4.
5.6. Kolby próżniowe, 500 ml.
5.7. Wodna pompa próżniowa.
5.8. Termometr kalibrowany od 0 do 100°C.
5.9. Eksykator z odpowiednim środkiem odwadniającym i wskaźnikiem wilgotności, np. żelem krzemionkowym lub równorzędnym.
5.10. Piec do suszenia z temperaturą uregulowaną na 150+/–2°C.
5.11. Pehametr.
5.12. Ogrzewana płytka.
5.13. Bibuła filtracyjna firmy Whatman nr 4 lub jej odpowiednik.
6. PROCEDURA
6.1. Do zlewki o pojemności 400 ml zważyć od 5 do 10 g (M gramów) analizowanej próbki, zawierającej ok. 50 do 100 mg cynku, dodać 50 ml wody destylowanej i wymieszać.
6.1.1. Przefiltrować, jeżeli jest to konieczne, za pomocą pompy próżniowej, zachować filtrat.
6.1.2. Powtórzyć czynność ekstrakcji z kolejnymi 50 ml wody destylowanej. Przefiltrować i połączyć filtraty.
6.2. Dla każdych 10 mg cynku, znajdujących się w roztworze (6.1.2), dodać 2 ml roztworu 2-metylochinolin-8-olu (4.7) i wymieszać.
6.3. Rozcieńczyć mieszaninę 150 ml wody destylowanej, doprowadzić mieszaninę do temperatury 60°C (5.12) i dodać 45 ml 0,2 M roztworu octanu amonu (4.6), stale mieszając.
6.4. Doprowadzić pH roztworu do 5,7–5,9: dodawać, mieszając, 6% roztwór amoniaku (4.5), pH roztworu zmierzyć pehametrem.
6.5. Odstawić roztwór na 30 minut. Przesączyć za pomocą wodnej pompy próżniowej przez tygiel z filtrem G-4, który wysuszono uprzednio (150°C) i zważono po schłodzeniu (M0 gramów), i przemyć osad 150 ml wody destylowanej o temperaturze 95°C.
6.6. Umieścić tygiel z filtrem w piecu suszącym o temperaturze nastawionej na 150°C i suszyć w ciągu jednej godziny.
6.7. Wyjąć filtr z pieca, umieścić w eksykatorze (5.9) i po schłodzeniu do temperatury pokojowej oznaczyć masę (M1 gramów).
7. OBLICZENIE
Obliczyć zawartość cynku w próbce jako procent wagowy (% m/m) za pomocą następującego wzoru:
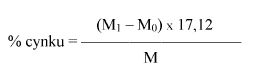
gdzie:
M = masa w gramach próbki pobranej do analizy według pkt 6.1,
M0 = masa w gramach pustego i suchego tygla z filtrem (6.5),
M1 = masa w gramach tygla z filtrem i osadem (6.7).
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości cynku ok. 1% (m/m) różnica między wynikami dwu równoległych oznaczeń, wykonywanych dla tej samej próbki, nie powinna przekraczać bezwzględnej wartości 0,1%.
VII. IDENTYFIKOWANIE I OZNACZANIE ZAWARTOŚCI KWASU 4-HYDROKSYBENZENOSULFONOWEGO
1. CEL I ZAKRES METODY
Metoda jest odpowiednia do identyfikowania i oznaczania kwasu 4-hydroksybenzenosulfonowego (INCI: 4-hydroxybenzenesulphonic acid) w produktach kosmetycznych, takich jak wyroby aerozolowe i płyny do twarzy.
2. DEFINICJA
Zawartość kwasu 4-hydroksybenzenosulfonowego oznaczona zgodnie z metodą jest wyrażona jako procent wagowy bezwodnego 4-hydroksybenzenosulfonianu cynku w wyrobie.
3. ZASADA
Próbka analityczna zostaje zatężona pod zmniejszonym ciśnieniem, rozpuszczona w wodzie i oczyszczona przez ekstrakcję chloroformem. Kwas 4-hydroksybenzenosulfonowy oznacza się jodometrycznie na części przesączonego roztworu wodnego.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. 36% (m/m) stężony kwas chlorowodorowy, d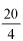
4.2. Chloroform.
4.3. Butan-1-ol.
4.4. Kwas octowy lodowaty.
4.5. Jodek potasu.
4.6. Bromek potasu.
4.7. Węglan sodu.
4.8. Kwas sulfanililowy (kwas 4-aminobenzenosulfonowy).
4.9. Azotan (III) sodu.
4.10. 0,1 N bromian (V) potasu.
4.11. 0,1 roztwór tiosiarczanu (VI) sodu.
4.12. 1% (m/v) wodny roztwór skrobi.
4.13. 2% (m/v) wodny roztwór węglanu sodu.
4.14. 4,5% (m/v) wodny roztwór azotanu (III) sodu.
4.15. 0,05% (m/v) roztworu ditizonu w chloroformie.
4.16. Roztwór rozwijający: butan-1-ol / kwas octowy lodowaty / woda (4:1:5 części objętościowych); po zmieszaniu w rozdzielaczu oddzielić dolną fazę.
4.17. Odczynnik Pauly'ego
Rozpuścić 4,5 g kwasu sulfanililowego (4.8) w 45 ml stężonego kwasu chlorowodorowego (4.1), ogrzewając, i rozcieńczyć roztwór wodą do 500 ml. Schłodzić 10 ml roztworu w naczyniu z wodą i lodem i dodać, mieszając, 10 ml zimnego roztworu azotanu (III) sodu (4.14). Odstawić roztwór na 15 minut w temperaturze 0°C (w tej temperaturze roztwór jest trwały w ciągu jednego do trzech dni) i bezpośrednio przed spryskiwaniem (7.5) dodać 20 ml roztworu węglanu sodu (4.13).
4.18. Gotowe przygotowane płytki celulozowe do chromatografii cienkowarstwowej: wielkość 20 x 20 cm, grubość warstwy adsorbenta 0,25 mm.
5. APARATURA I WYPOSAŻENIE
5.1. Okrągłodenne kolby ze szlifowanym korkiem szklanym, 100 ml.
5.2. Rozdzielacz, 100 ml.
5.3. Kolba stożkowa ze szlifowanym korkiem szklanym, 250 ml.
5.4. Biureta, 25 ml.
5.5. Pipety ze zbiornikiem gruszkowym, 1, 2 i 10 ml.
5.6. Pipeta kalibrowana, 5 ml.
5.7. Strzykawka, 10 µl z kalibracją co 0,1 µl.
5.8. Termometr ze skalą od 0 do 100°C.
5.9. Łaźnia wodna z elementem grzewczym.
5.10. Piec do suszenia, dobrze przewietrzany i nastawiony na temperaturę 80°C.
5.11. Typowy aparat do chromatografii cienkowarstwowej.
6. PRZYGOTOWANIE PRÓBKI
W opisanej poniżej metodzie identyfikacji i oznaczania kwasu hydroksybenzenosulfonowego w wyrobach aerozolowych używana jest pozostałość otrzymana przez usunięcie z pojemnika aerozolowego rozpuszczalników i propelentów, które wyparowują pod normalnym ciśnieniem.
7. IDENTYFIKOWANIE
7.1. Nanieść za pomocą strzykawki (5.7) po 5 µl pozostałości (6) lub próbki w każdym z sześciu punktów na linii początkowej w odległości 1 cm od dolnej krawędzi płytki do chromatografii cienkowarstwowej (4.18).
7.2. Umieścić płytkę w komorze rozwijającej, która już zawiera roztwór rozwijający (4.16), i rozwijać do osiągnięcia przez czoło rozpuszczalnika odległości 15 cm od linii początkowej.
7.3. Wyjąć płytkę z roztworu rozwijającego i suszyć w temperaturze 80°C do momentu, kiedy opary kwasu octowego nie będą wyczuwalne. Spryskiwać płytkę roztworem węglanu sodu (4.13) i wysuszyć na powietrzu.
7.4. Przykryć połowę płytki szklaną płytką i spryskać nieprzykrytą część 0,05% roztworem ditizonu (4.15). Pojawienie się purpurowo-czerwonych plam na chromatogramie wskazuje na obecność jonów cynku.
7.5. Przykryć spryskaną część płytki szklaną płytką i spryskać pozostałą część odczynnikiem Pauly'ego (4.17). Na obecność kwasu 4-hydroksybenzenosulfonowego wskazuje pojawienie się żółtobrązowych plam o wartości Rf ok. 0,26, podczas gdy żółta plama o wartości Rf ok. 0,45 na chromatogramie wskazuje na obecność kwasu 3-hydroksybenzenosulfonowego.
8. OZNACZENIE
8.1. Do okrągłodennej kolby o pojemności 100 ml odważyć 10 g próbki lub pozostałości (6) i odparować prawie do sucha pod próżnią w wyparce obrotowej w łaźni wodnej o temperaturze 40°C.
8.2. Wprowadzić pipetą do kolby 10,0 ml (V1 ml) wody i rozpuścić pozostałość po odparowaniu (8.1) przez ogrzewanie.
8.3. Ilościowo przenieść roztwór do rozdzielacza (5.2) i ekstrahować roztwór wodny dwukrotnie porcjami po 20 ml chloroformu (4.2). Po każdej ekstrakcji odrzucać fazę chloroformową.
8.4. Przesączyć roztwór wodny przez karbowany sączek. Zależnie od oczekiwanej zawartości kwasu hydroksybenzenosulfonowego przenieść pipetą 1,0 lub 2,0 ml (V2) przesączu do 250 ml kolby stożkowej (5.3) i rozcieńczyć wodą do 75 ml.
8.5. Dodać 2,5 ml 36% kwasu chlorowodorowego (4.1) i 2,5 g bromku potasu (4.6), zmieszać i doprowadzić temperaturę roztworu do temperatury 50°C za pomocą łaźni wodnej.
8.6. Dodawać z biurety 0,1 N roztworu bromianu (V) potasu (4.10), aż roztwór, utrzymywany w temperaturze 50°C, stanie się żółty.
8.7. Dodać dalsze 3,0 ml roztworu bromianu (V) potasu (4.10), zamknąć kolbę korkiem i pozostawić na 10 minut w łaźni wodnej o temperaturze 50°C.
Jeżeli po 10 minutach roztwór straci swoją barwę, dodać dalsze 2,0 ml roztworu bromianu (V) potasu (4.10), zamknąć kolbę korkiem i ogrzewać przez 10 minut w łaźni wodnej utrzymywanej w temperaturze 50°C. Zanotować całkowitą ilość dodanego roztworu bromianu (V) potasu (a).
8.8. Schłodzić roztwór do temperatury pokojowej, dodać 2 g jodku potasu (4.5) i wymieszać.
8.9. Odmiareczkować powstały jod 0,1 N roztworem tiosiarczanu (VI) sodu (4.11). Przy końcu miareczkowania dodać kilka kropli roztworu skrobi (4.12) jako wskaźnika. Zanotować ilość użytego tiosiarczanu sodu (b).
9. OBLICZENIE
Obliczyć zawartość hydroksybenzenosulfonianu cynku w próbce lub pozostałości (6) jako procent masowy (m/m) za pomocą następującego wzoru:

gdzie:
a = całkowita ilość w mililitrach dodanego 0,1 N roztworu bromianu (V) potasu (8.7),
b = ilość w mililitrach 0,1 N roztworu tiosiarczanu (VI) sodu użytego do odmiareczkowania (8.9),
m = ilość analizowanego produktu lub pozostałości wyrażona w miligramach (8.1),
V1 = objętość roztworu otrzymanego według pkt 8.2 wyrażona w mililitrach,
V2 = objętość rozpuszczonej pozostałości po odparowaniu użytej do analizy (8.4) wyrażona w mililitrach.
Uwaga: W przypadku wyrobów aerozolowych wynik pomiaru w % (m/m) pozostałości (6) musi być wyrażony w odniesieniu do oryginalnego wyrobu. W celu wykonania tej zamiany podano zasady pobierania próbek aerozoli.
10. POWTARZALNOŚĆ (patrz: norma ISO/DIS 5725)
Dla zawartości ok. 5% hydroksybenzenosulfonianu cynku różnica między wynikami dwu oznaczeń, przeprowadzonych równolegle dla tej samej próbki, nie powinna przekraczać bezwzględnej wartości 0,5%.
11. Maksymalna zawartość 4-hydroksybenzenosulfonianu cynku w dezodorantach, produktach przeciwpotowych i płynach ściągających wynosi 6% w przeliczeniu na substancję bezwodną. Sformułowanie to oznacza, że obok zawartości kwasu hydroksybenzenosulfonowego należy oznaczyć zawartość cynku. Pomnożenie obliczonej zawartości hydroksybenzenosulfonianu cynku (9) przez współczynnik 0,1588 daje minimalną zawartość cynku w % (m/m), która teoretycznie musi się znajdować w wyrobie w związku z oznaczoną zawartością kwasu hydroksybenzenosulfonowego. Zawartość cynku – rzeczywiście oznaczona grawimetrycznie – może jednakże być wyższa, ponieważ chlorek cynku i siarczan cynku mogą być również używane w produktach kosmetycznych.
VIII. IDENTYFIKCJA ŚRODKÓW UTLENIAJĄCYCH I OZNACZANIE NADTLENKU WODORU W PRODUKTACH KOSMETYCZNYCH DO PIELĘGNACJI WŁOSÓW
CEL I ZAKRES
Jodometryczne oznaczanie nadtlenku wodoru w produktach kosmetycznych jest możliwe wyłącznie w przypadku nieobecności innych środków utleniających, które uwalniają jod z jodków. W związku z tym przed jodometrycznym oznaczeniem nadtlenku wodoru, są konieczne wykrycie i identyfikacja innych występujących środków utleniających. Identyfikacja dzieli się na dwa etapy: pierwszy obejmuje nadsiarczany, bromiany i nadtlenek wodoru, a drugi nadtlenek baru.
A. IDENTYFIKCJA NADSIARCZANÓW, BROMIANÓW I NADTLENKU WODORU
1. ZASADA
Identyfikację nadsiarczanu sodu, nadsiarczanu potasu i nadsiarczanu amonu oraz bromianu potasu, bromianu sodu i nadtlenku wodoru – nawet tych pochodzących z nadtlenku baru – wykonuje się za pomocą chromatografii bibułowej zstępującej, z zastosowaniem dwóch rozpuszczalników rozwijających.
2. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy. 2.1. Wodne roztwory o odniesieniu 0,5% (m/v) następujących związków:
2.1.1. Nadsiarczan sodu.
2.1.2. Nadsiarczan potasu.
2.1.3. Nadsiarczan amonu.
2.1.4. Bromian potasu.
2.1.5. Bromian sodu.
2.1.6. Nadtlenek wodoru.
2.2. Rozpuszczalnik rozwijający A, etanol 80% (v/v).
2.3. Rozpuszczalnik rozwijający B, benzen / metanol / 3-metylobutan-1-ol / woda (34:38:18:10 obj.).
2.4. Środek wywołujący A, 10% (m/v) roztwór wodny jodku potasu.
2.5. Środek wywołujący B, 1% (m/v) roztwór wodny skrobi.
2.6. Środek wywołujący C, 10% (m/m) kwas chlorowodorowy.
2.7. 4 N kwas chlorowodorowy.
3. APARATURA I WYPOSAŻENIE
3.1. Bibuła chromatograficzna (bibuła Whatman nr 3 i nr 4 lub jej równoważniki).
3.2. Mikropipeta, 1 µl.
3.3. Kolby miarowe, 100 ml.
3.4. Sączki karbowane.
3.5. Aparatura do zstępującej chromatografii bibułowej.
4. PRZYGOTOWANIE PRÓBKI
4.1. Produkty rozpuszczalne w wodzie
Przygotować po dwa roztwory każdej próbki, przez rozpuszczenie odpowiednio 1 g i 5 g produktu w 100 ml wody. Pobrać 1 µl każdego z tych roztworów do analizy metodą chromatografii bibułowej, opisaną w pkt 5.
4.2. Produkty trudno rozpuszczalne w wodzie
4.2.1. Odważyć 1 g i 5 g próbki i zawiesić w 50 ml wody, uzupełnić do 100 ml wodą w obu przypadkach i wymieszać. Przesączyć obie dyspersje przez sączek karbowany (3.4) i pobrać po 1 µl każdego filtratu do analizy metodą chromatografii bibułowej, opisaną w pkt 5.
4.2.2. Przygotować ponownie dwie dyspersje każdej próbki przez dyspergowanie 1 g i 5 g w 50 ml wody, zakwaszając rozwodnionym kwasem solnym (2.7), mieszając i uzupełniając do 100 ml wodą. Przesączyć dyspersję przez sączek karbowany i pobrać 1 µl każdego filtratu do analizy metodą chromatografii bibułowej opisaną w pkt 5.
4.3. Kremy
Zawiesić 5 g i 20 g każdego produktu w 100 ml wody i zastosować dyspersje do wykonania analizy metodą chromatografii bibułowej opisaną w pkt 5.
5. METODA
5.1. Umieścić właściwą ilość rozpuszczalników A (2.2) i B (2.3) w komorach dwóch oddzielnych chromatografów w celu wykonania zstępującej chromatografii bibułowej. Nasycać komory chromatograficzne oparami rozpuszczalników przez co najmniej 24 godziny.
5.2. Nanieść po 1 µl jednego roztworu próbki i jednego roztworu odniesienia, przygotowanych według opisu w pkt 4 i 2.1, na każdy punkt startowy paska bibuły chromatograficznej (Whatman nr 3 lub równoważnej) o długości 40 cm i szerokości 20 cm (3.1), lub innego odpowiedniego formatu, i odparować rozpuszczalnik na powietrzu.
5.3. Umieścić pasek chromatograficzny (5.2) w komorze chromatograficznej z rozpuszczalnikiem rozwijającym A (5.1) i rozwijać do chwili, kiedy czoło rozpuszczalnika osiągnie odległość 35 cm (ok. 15 godzin).
5.4. Powtórzyć procedurę opisaną w pkt 5.2 i 5.3, z użyciem bibuły chromatograficznej (Whatman nr 4 lub równoważnej) (3.1) i rozpuszczalnika rozwijającego B (2.3). Rozdział chromatograficzny prowadzić do chwili, kiedy czoło rozpuszczalnika osiągnie odległość 35 cm (ok. pięciu godzin).
5.5. Po rozwinięciu wyjąć chromatogramy z komory i wysuszyć na powietrzu.
5.6. Wywołać plamy na chromatogramie przez spryskanie go kolejno:
5.6.1. Środkiem wywołującym A (2.4) i następnie, po krótkim czasie, środkiem wywołującym B (2.5). Plamy nadsiarczanów pojawiają się na chromatografie najpierw, a następnie zostaną wywołane plamy nadtlenku wodoru. Obrysować plamy ołówkiem.
5.6.2. Środkiem wywołującym C (2.6) chromatogramów otrzymanych zgodnie z opisem w pkt 5.6.1; szaroniebieskie plamy na chromatogramie wskazują na obecność bromianów.
5.7. W opisanych powyżej warunkach, właściwych dla rozpuszczalników rozwijających A (2.2) i B (2.3), wartości Rf dla substancji odniesienia (2.1) są w przybliżeniu następujące:
|
| Roztwór rozwijający A (2.2) | Roztwór rozwijający B (2.3) |
| Nadsiarczan sodu | 0,40 | 0,10 |
| Nadsiarczan potasu | 0,40 | 0,02 + 0,05 |
| Nadsiarczan amonu | 0,50 | 0,10 + 0,20 |
| Bromian sodu | 0,40 | 0,20 |
| Bromian potasu | 0,40 | 0,10 + 0,20 |
| Nadtlenek wodoru | 0,80 | 0,80 |
B. IDENTYFIKACJA NADTLENKU BARU
1. ZASADA
Identyfikację nadtlenku baru wykonuje się na podstawie powstawania nadtlenku wodoru, po zakwaszeniu próbki (A.4.2), oraz na podstawie stwierdzenia obecności jonu baru:
1) w przypadku nieobecności nadsiarczanów (A), przez dodanie rozcieńczonego kwasu siarkowego do części kwaśnego roztworu próbki (4.1), w wyniku czego powstaje biały osad siarczanu baru. Obecność jonów baru w próbce (4.1) potwierdza się ponownie za pomocą chromatografii bibułowej według metody opisanej poniżej (5);
2) w przypadku, gdy w próbce są obecne równocześnie nadtlenek baru i nadsiarczany (4.2), obecność jonów baru w roztworze stopu (4.2.3) stwierdza się przez rozpuszczenie pozostałości z roztworu (4.2) w podwyższonej temperaturze i w środowisku alkalicznym, po rozpuszczeniu w kwasie chlorowodorowym, za pomocą metody chromatografii bibułowej i/lub przez wytrącenie osadu siarczanu baru.
2. ODCZYNNIKI
2.1. Metanol.
2.2. 36% (m/m) stężony kwas chlorowodorowy.
2.3. 6 N kwas chlorowodorowy.
2.4. 4 N kwas siarkowy.
2.5. Sól dwusodowa kwasu rodyzonowego (3,4,5,6-tetraoksocyklohekseno-1,2-diolu).
2.6. Chlorek baru (BaCl2 2H2O).
2.7. Bezwodny węglan sodu.
2.8. 1% (m/v) roztwór wodny chlorku baru.
2.9. Rozpuszczalnik rozwijający zawierający metanol, stężony kwas chlorowodorowy (stężenie 36%) i wodę (80:10:10 obj.).
2.10. Środek wywołujący, 0,1% (m/v) roztwór wodny soli dwusodowej kwasu rodyzonowego, świeżo przygotowany bezpośrednio przez użyciem.
3. APARATURA I WYPOSAŻENIE
3.1. Mikropipeta, 5 µl.
3.2. Tygle platynowe.
3.3. Kolby miarowe, 100 ml.
3.4. Bibuła chromatograficzna firmy Schleicher and Schüll 2043b lub równoważna. Bibułę oczyszcza się przez pozostawienie jej na noc w komorze chromatograficznej (A.3.5) zawierającej rozpuszczalnik rozwijający (2.9), a następnie suszy się ją.
3.5. Karbowany sączek bibułowy.
3.6. Aparatura do zstępującej chromatografii bibułowej.
4. PRZYGOTOWANIE PRÓBKI
4.1. Produkty niezawierające nadsiarczanów
4.1.1. Zawiesić 2 g produktu w 50 ml wody i doprowadzić kwasem chlorowodorowym (2.3) pH dyspersji do wartości ok. 1.
4.1.2. Przenieść dyspersję za pomocą wody do 100 ml kolby miarowej, uzupełnić wodą do kreski i wymieszać. Dyspersji używa się do analizy metodą chromatografii bibułowej opisanej w pkt 5 oraz do identyfikacji baru przez wytrącenie siarczanu.
4.2. Produkty zawierające nadsiarczany
4.2.1. Zawiesić 2 g produktu w 100 ml wody i przesączyć.
4.2.2. Do wysuszonej pozostałości dodać siedem do dziesięciu razy więcej wagowo węglanu sodu (2.7), wymieszać i stapiać mieszaninę w tyglu platynowym (3.2) przez pół godziny.
4.2.3. Schłodzić do temperatury pokojowej, rozpuścić stop w 50 ml wody i przesączyć (3.5).
4.2.4. Rozpuścić pozostałość ze stopu w kwasie chlorowodorowym (2.3) i uzupełnić wodą do 100 ml. Roztworu tego używa się do analizy metodą chromatografii bibułowej, opisaną w pkt 5, oraz do identyfikowania obecności baru przez wytrącenie siarczanu.
5. METODA
5.1. Umieścić właściwą ilość rozpuszczalnika rozwijającego (2.9) w komorze do zstępującej chromatografii bibułowej i nasycać komorę przez co najmniej 15 godzin.
5.2. Na kawałku bibuły chromatograficznej – przygotowanej tak, jak opisano w pkt 3.4 – nanieść po 5 µl każdego roztworu, przygotowanego zgodnie ze wskazówkami przedstawionymi w pkt 4.1.2 i 4.2.4, oraz roztwór odniesienia (2.8) w trzech punktach startowych.
5.3. Wysuszyć bibułę z naniesionymi próbkami i roztworem odniesienia na powietrzu. Rozwijać chromatogram do chwili, kiedy czoło rozpuszczalnika osiągnie 30 cm.
5.4. Wyjąć chromatogram z komory i wysuszyć na powietrzu.
5.5. Wywołać plamy na chromatogramie przez spryskanie bibuły środkiem wywołującym (2.10). W przypadku obecności baru na chromatogramie pojawiają się czerwone plamy o wartości Rf ok. 0,10.
C. OZNACZENIE NADTLENKU WODORU
1. ZASADA
Jodometryczne oznaczanie nadtlenku wodoru przeprowadza się według następującej reakcji:
H2O2 + 2H+ + 2I- 
Przemiana zachodzi powoli, lecz można ją przyspieszyć przez dodanie heptamolibdenianu (VI) amonu. Ilość powstałego jodu oznacza się miareczkowo tiosiarczanem sodu i stanowi ona miarę zawartości nadtlenku wodoru.
2. DEFINICJA
Zawartość nadtlenku wodoru oznaczona w sposób opisany poniżej jest wyrażona jako procent masowy (% m/m) produktu.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. 2 N kwas siarkowy.
3.2. Jodek potasu.
3.3. Heptamolibdenian (VI) amonu.
3.4. 0,1 N tiosiarczan sodu.
3.5. 10% (m/v) roztwór jodku potasu, świeżo przygotowany bezpośrednio przez użyciem.
3.6. 20% (m/v) roztwór heptamolibdenianu (VI) amonu.
3.7. 1% (m/v) roztwór skrobi.
4. APARATURA I WYPOSAŻENIE
4.1. Zlewki, 100 ml.
4.2. Biureta, 50 ml.
4.3. Kolby miarowe, 250 ml.
4.4. Cylindry miarowe, 25 i 100 ml.
4.5. Pipety jednomiarowe, 10 ml.
4.6. Kolby stożkowe, 250 ml.
5. METODA
5.1. W zlewce 100 ml odważyć ok. 10 g (m gramów) produktu zawierającego ok. 0,6 g nadtlenku wodoru. Przenieść zawartość zlewki za pomocą wody do kolby miarowej 250 ml, uzupełnić wodą do kreski i wymieszać.
5.2. Odmierzyć pipetą 10 ml roztworu próbki (5.1) do 250 ml kolby stożkowej (4.6) i dodawać kolejno 100 ml 2N kwasu siarkowego (3.1), 20 ml roztworu jodku potasu (3.5) i trzy krople roztworu heptamolibdenianu (VI) amonu (3.6).
5.3. Miareczkować powstały jod bezzwłocznie 0,1 N roztworem tiosiarczanu sodu (3.4) i bezpośrednio przed osiągnięciem punktu końcowego dodać kilka mililitrów roztworu skrobi (3.7) jako wskaźnika. Zarejestrować zużycie 0,1 N roztworu tiosiarczanu sodu (3.4) w mililitrach (V).
5.4. Przeprowadzić analizę ślepej próby według sposobu opisanego w pkt 5.2 i 5.3, zastępując 10 ml roztworu próbki 10 ml wody. Zarejestrować zużycie 0,1 N roztworu tiosiarczanu sodu w analizie ślepej próby (Vo ml).
6. OBLICZENIE
Obliczyć zawartość nadtlenku wodoru w produkcie, w procentach masowych (% m/m), według następującego wzoru:
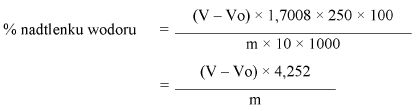
gdzie:
m = ilość produktu poddawanego analizie (5.1),
Vo = zużycie 0,1 N roztworu tiosiarczanu do analizy ślepej próby w mililitrach (5.4),
V = zużycie 0,1 N roztworu tiosiarczanu do analizy roztworu próbki w mililitrach (5.3).
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
W przypadku produktu zawierającego ok. 6% m/m nadtlenku wodoru różnica pomiędzy wynikami dwóch oznaczeń, przeprowadzonych równolegle dla tej samej próbki, nie powinna przekraczać wartości bezwzględnej 0,2%.
IX. IDENTYFIKACJA I PÓŁILOŚCIOWE OZNACZANIE NIEKTÓRYCH BARWNIKÓW UTLENIAJĄCYCH W FARBACH DO WŁOSÓW
1. CEL I ZAKRES
Metoda jest odpowiednia do identyfikacji i półilościowego oznaczania następujących substancji w farbach do włosów w postaci kremu lub płynu:
| Substancje | Symbol |
| Fenylenodwuaminy |
|
| o-fenylenodwuamina | (OPD) |
| m-fenylenodwuamina | (MPD) |
| p-fenylenodwuamina | (PPD) |
| Metylofenylenodwuaminy |
|
| 4-metylo-1,2-fenylenodwuamina (3,4-dwuaminotoluen) | (OTD) |
| 4-metylo-1,3-fenylenodwuamina (2,4-dwuaminotoluen) | (MTD) |
| 2-metylo-1,4-fenylenodwuamina (2,5 dwuaminotoluen) | (PTD) |
| Dwuaminofenole |
|
| 2,4-dwuaminofenol | (DAP) |
| Hydrochinon |
|
| 1,4-dwuhydroksybenzen | (H) |
| a-naftol | (a-N) |
| Pirogalol |
|
| 1,2,3-trójhydroksybenzen | (P) |
| Rezorcynol |
|
| 1,3-dwuhydroksybenzen | (R) |
2. ZASADA
Barwniki utleniające z farb w postaci kremu lub płynu ekstrahuje się za pomocą 96% alkoholu etylowego oraz przy pH = 10 i identyfikuje się je metodą chromatografii cienkowarstwowej, jedno- lub dwukierunkowej.
W celu półilościowego oznaczenia tych substancji chromatogramy próbek otrzymane za pomocą czterech układów rozwijających są porównywane z chromatogramami substancji odniesienia otrzymanymi w tym samym czasie i w możliwie jak najbardziej podobnych warunkach.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. Bezwodny alkohol etylowy.
3.2. Aceton.
3.3. Alkohol etylowy, 96% v/v.
3.4. Roztwór amoniaku, 25%, d 
3.5. Kwas L(+) askorbinowy.
3.6. Chloroform.
3.7. Cykloheksan.
3.8. Azot techniczny.
3.9. Toluen.
3.10. Benzen.
3.11. n-butanol.
3.12. Butan-2-ol.
3.13. Kwas podfosforawy, roztwór 50% v/v.
3.14. Odczynnik do dwuazowania. Jeden z dwóch do wyboru:
1) chlorobenzenosulfonian3-nitro-1-benzenodwuazoniowy (stabilizowany w postaci soli), tak jak w Red 2 JN-Francolor;
2) naftalenobenzoesan-2-chloro-4-nitro-1-benzenodwuazoniowy (stabilizowany w postaci soli), tak jak w odczynniku NNCD – odniesienie nr 74150 FLUKA
lub równoważny.
3.15. Azotan srebra.
3.16. p-dwumetyloaminobenzaldehyd.
3.17. 2,5-dwumetylofenol.
3.18. Sześciowodny chlorek żelazowy.
3.19. Kwas chlorowodorowy, roztwór 10% m/v.
3.20. Substancje odniesienia
Substancje odniesienia przedstawiono w pkt 1 „Cel i zakres”. W przypadku związków aminowych substancją odniesienia może być chlorowodorek (mono- lub di-) lub wolna zasada.
3.21. Roztwory odniesienia 0,5% (m/v)
Przygotowuje się roztwór A 0,5% (m/v) wszystkich substancji odniesienia podanych w pkt 3.20.
Odważyć 50 mg +/– 1 mg substancji odniesienia w 10 ml kolbie miarowej.
Dodać 5 ml 96% alkoholu etylowego (3.3) i 250 mg kwasu askorbinowego (3.5).
Zalkalizować, dodając roztwór amoniaku (3.4), do uzyskania wyraźnego odczynu o pH 10 (zbadać papierkiem wskaźnikowym).
Uzupełnić 96% alkoholem etylowym (3.3) do 10 ml i wymieszać.
Roztwory można przechowywać przez tydzień w chłodnym miejscu i bez dostępu światła.
W niektórych przypadkach po dodaniu kwasu askorbinowego i amoniaku może wytrącać się osad. W tej sytuacji przed przystąpieniem do kolejnych czynności należy pozwolić na jego osadzenie się.
3.22. Rozpuszczalniki rozwijające.
3.22.1. Aceton / chloroform / toluen (35:25:40 obj.).
3.22.2. Chloroform / cykloheksan / etanol absolutny / 25% amoniak (80:10:10:1 obj.).
3.22.3. Benzen / butan-2-ol / woda (50:25:25). Po rozdzieleniu w temperaturze pokojowej (od 20 do 25°C) należy dobrze wstrząsnąć i zastosować górną fazę.
3.22.4. Butanol / chloroform / odczynnik M (7:70:23 obj.). Rozdzielać starannie w temperaturze pokojowej (od 20 do 25°C) i stosować dolną fazę. Przygotowanie odczynnika M
| 25% (v/v) roztwór amoniaku | 24 objętości |
| 50% kwas podfosforawy (3.13) | l objętość |
| Woda | 75 objętości |
Uwaga: Rozpuszczalniki rozwijające zawierające amoniak bezpośrednio przed użyciem należy dobrze wstrząsać.
3.23. Wskaźniki wywołujące do rozpylania.
3.23.1. Odczynnik do dwuazowania
Przygotować 5% (m/v) roztwór wodny wybranego odczynnika (3.14). Roztwór należy przygotować bezpośrednio przed użyciem.
3.23.2. Odczynnik Ehrlicha
Rozpuścić 2 g p-dwumetyloaminobenzaldehydu (3.16) w 100 ml 10% (m/v) wodnego roztworu kwasu chlorowodorowego (3.19).
3.23.3. 2,5-dwumetylofenol / chlorek żelaza sześciowodny
Roztwór 1: rozpuścić 1 g dwumetylofenolu (3.17) w 100 ml 96% etanolu (3.3)
Roztwór 2: rozpuścić 4 g chlorku żelaza sześciowodnego (3.18) w 100 ml 96% etanolu (3.3).
Do wywoływania roztwory należy rozpylać oddzielnie, najpierw roztwór 1, następnie roztwór 2.
3.23.4. Amoniakalny azotan srebra
Do 5% (m/v) wodnego roztworu azotanu srebra (3.15) dodać 25% amoniak (3.4) aż do rozpuszczenia osadu. Odczynnik ten musi zostać przygotowany bezpośrednio przed użyciem. Nie należy go przechowywać. 4. APARATURA
4.1. Stosuje się zwykłe wyposażenie laboratoryjne do chromatografii cienkowarstwowej
4.1.1. Osłona z tworzywa sztucznego lub szklana, skonstruowana tak, aby azot mógł opływać płytkę chromatograficzną podczas nanoszenia kropli próbek i suszenia. Ostrożność ta jest konieczna ze względu na podatność pewnych barwników na utlenianie.
4.1.2. Mikrostrzykawka 10 µl, kalibrowana, z podziałkami 0,2 µl, z okrągło zakończoną igłą, lub lepiej 50 µl wielokrotny dozownik, zmontowany na statywie śrubowym w taki sposób, że płytka może być utrzymywana w atmosferze azotu.
4.1.3. Cienkowarstwowe płytki krzemionkowe gotowe do stosowania, o grubości 0,25 mm, o wymiarach 20 x 20 cm, firmy Macherey and Nagel, Silica G-HR, które mają podłoże ze sztucznego tworzywa, lub płytki równoważne.
4.2. Wirówka, 4000 obrotów/minutę.
4.3. Probówki do wirówki, 10 ml, z korkiem gwintowym pokrytym PTFE lub probówki równoważne.
5. PROCEDURA
5.1. Przygotowanie próbek
Odrzucić pierwsze 2 lub 3 cm kremu wyciśniętego z tuby.
W probówce wirówki (4.3), uprzednio przepłukanej azotem, umieścić: 300 mg kwasu askorbinowego, 3 g kremu lub 3 g homogenizowanego płynu.
Wkraplać 25% amoniak (3.4) do uzyskania pH 10. Uzupełnić 96% alkoholem etylowym (3.3) do 10 ml.
Homogenizować w atmosferze azotu (3.8), zamknąć i następnie wirować przy 4000 obrotów na minutę przez 10 minut.
Do analizy stosować ciecz znajdującą się na górze.
5.2. Chromatografia
5.2.1. Nanoszenie kropli na płytki
W atmosferze azotu (3.8) nanieść na płytkę chromatograficzną (4.1.3) po 1 µl wszystkich opisanych powyżej roztworów odniesienia, w dziewięciu punktach, oddalonych od siebie o 1,5 cm, wzdłuż linii znajdującej się średnio 1,5 cm od krawędzi płytki. Roztwory odniesienia należy rozmieścić następująco:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| R | P | H | PPD | DAP | PTD | OPD | OTD | MPD |
| MTD | a-N |
|
|
|
|
|
|
|
Dodatkowo nanieść w pkt 10 i 11 odpowiednio po 2 µl próbek badanych roztworów, otrzymanych zgodnie z opisem w pkt 5.1.
Utrzymywać płytkę w atmosferze azotu aż do chwili, kiedy zostanie poddana rozdziałowi chromatograficznemu.
5.2.2. Rozwijanie
Umieścić płytkę w komorze uprzednio przepłukanej azotem (3.8), nasyconej jednym z czterech rozpuszczalników (3.22) i pozostawić do rozwijania w temperaturze pokojowej (od 20 do 25°C), w ciemności, do chwili, kiedy czoło rozpuszczalnika osiągnie wysokość ok. 15 cm od linii bazowej. Wyjąć płytkę z komory i suszyć w atmosferze azotu (3.8), w temperaturze pokojowej.
5.2.3. Spryskiwanie
Spryskać płytkę bezzwłocznie jednym z czterech roztworów wymienionych w pkt 3.23.
5.2.4. Identyfikacja
Porównać wartości Rf i barwy otrzymanych plam z wartościami i barwami plam otrzymanymi dla substancji odniesienia poddanej chromatograficznemu rozdziałowi. W tabeli I podano przykłady wartości Rf i barwy dla każdej substancji zależnie od stosowanych rozpuszczalników i użytych wskaźników wywołujących. Potwierdzenie wątpliwej identyfikacji można czasem uzyskać metodą wzorca wewnętrznego, dodając roztwór odpowiedniej substancji odniesienia do ekstraktu próbki.
5.2.5. Ocena półilościowa
Porównać wizualnie intensywność plam dla każdej substancji identyfikowanej zgodnie z pkt 5.2.4 z odpowiednim zakresem stężeń substancji odniesienia. Jeżeli stężenie jednej lub kilku substancji znalezionych w próbce jest zbyt wysokie, rozcieńczyć ekstrakt próbki i powtórzyć pomiar.
TABELA I
Wartości Rf i barwy otrzymane bezpośrednio po spryskaniu
| Substancja odniesienia (3.20) | Rozpuszczalniki rozwijające | Rozpylane wskaźniki wywołujące | ||||||
| wartości Rf | otrzymane barwy | |||||||
| (3.22.1) | (3.22.2) | (3.22.3) | (3.22.4) | dwuazowy (3.23.1) | Ehrlicha (3.23.2) | dwumetylofenol (3.23.3) | AgNO3 (3.24.4) | |
| OPD | 0,62 | 0,60 | 0,30 | 0,57 | blado-brązowa | – | – | blado-brązowa |
| MPD | 0,40 | 0,60 | 0,47 | 0,48 | fioletowo-brązowa* | żółta | blado-brązowa | blado-brązowa |
| PPD | 0,20 | 0,50 | 0,30 | 0.48 | brązowa | jasnoczerwona* | fioletowa | szara |
| OTO | 0,60 | 0,60 | 0,53 | 0,60 | brązowa* | blado-pomarańczowa | blado-brązowa | szarawo-brązowa |
| MTD | 0,40 | 0,67 | 0,45 | 0,60 | czerwonawo-brązowa* | żółta | brązowa | czarna |
| PTD | 0,33 | 0,65 | 0,37 | 0,70 | brązowa | pomarańczowa | fioletowa* | szara |
| DAP | 0,07 | – | 0 | 0,05 | brązowa* | pomarańczowa | fioletowa | brązowa |
| H | 0,50 | 0,35 | 0,80 | 0,20 | – | pomarańczowa | fioletowa | czarna* |
| a-N | 0,90 | 0,80 | 0,90 | 0,75 | pomarańczowo-brązowa | – | fioletowa* | czarna |
| P | 0,37 | – | 0,67 | 0,05 | brązowa | bardzo lekko blado-fioletowa | bardzo lekko blado-brązowa | brązowa* |
| R | 0,50 | 0,37 | 0,80 | 0,17 | pomarańczowa* | blado-fioletowa | bardzo lekko blado-brązowa | blado-brązowa |
| Uwaga: 1. OPD jest słabo widoczna, należy użyć rozpuszczalnika (3.22.3), aby rozdzielić ją wyraźnie od OTD. 2. * Wskazuje najlepszą wywołaną barwę. | ||||||||
6. BADANIE METODĄ DWUKIERUNKOWEJ CHROMATOGRAFII CIENKOWARSTWOWEJ
Procedura chromatografii dwukierunkowej wymaga zastosowania dodatkowych wzorców i odczynników.
6.1. Dodatkowe roztwory i substancje odniesienia
6.1.1. β-naftol (B-N).
6.1.2. 2-aminofenol (OAP).
6.1.3. 3-aminofenol (MAP).
6.1.4. 4-aminofenol (PAP).
6.1.5. 2-nitro-1,4-fenylenodwuamina (2-NPPD).
6.1.6. 4-nitro-1,2-fenylenodwuamina (4-NOPD).
Przygotować 0,5% m/v roztwory wszystkich dodatkowych substancji odniesienia według wskazówek przedstawionych w pkt 3.21.
6.2. Dodatkowy rozpuszczalnik rozwijający
Octan etylu / cykloheksan / 25% roztwór amoniaku (65:30:0,5 obj.).
6.3. Dodatkowy układ wskaźnika wywołującego
Umieścić szklane naczynie w komorze do chromatografii cienkowarstwowej, dodać ok. 2 g kryształów jodu i zamknąć komorę odpowiednią pokrywką.
6.4. Chromatografia
6.4.1. Na powierzchni absorbentu płytki cienkowarstwowej (4.1.3) narysować dwie linie, tak jak pokazano na rys. 6.
6.4.2. W atmosferze azotu (4.1.1) nanieść od 1 do 4 µl ekstraktu (5.1) w punkcie bazowym 1 (rys. 6), który znajduje się w odległości 2 cm od obu krawędzi. Ilość naniesionego ekstraktu zależy od intensywności plam na chromatogramach (5.2).
6.4.3. Między pkt 2 i 3 (rys. 6) rozdzielić barwniki utleniające, zidentyfikowane lub do identyfikacji, przez rozdział chromatograficzny (5.2) (odległość między punktami wynosi 1,5 cm). Nanosić po 2 µl wszystkich roztworów odniesienia, z wyjątkiem DAP, który trzeba nanieść w ilości 6 µl. Czynności należy wykonywać w atmosferze azotu (6.4.2).
6.4.4. Powtórzyć operację (6.4.3) w punktach bazowych 4 i 5 (rys. 6) i utrzymywać płytkę w atmosferze azotu aż do chwili rozpoczęcia rozdziału chromatograficznego (odległość między punktami wynosi 1,5 cm).
6.4.5. Przepłukać komorę chromatograficzną azotem (3.8) i umieścić w niej odpowiednią ilość rozpuszczalnika rozwijającego (3.22.2). Umieścić płytkę (6.4.4) w komorze i rozwinąć ją w ciemności w pierwszym kierunku elucji (rys. 6).
Eluować do chwili, kiedy czoło rozpuszczalnika osiągnie linię zaznaczoną na płytce (ok. 13 cm).
6.4.6. Wyjąć płytkę z komory i umieścić ją w komorze chromatograficznej przepłukanej uprzednio azotem, co najmniej na 60 minut, w celu odparowania wymywanego rozpuszczalnika.
6.4.7. Wprowadzić do komory przepłukanej azotem (3.8) odpowiednią ilość rozpuszczalnika wymywającego (6.2) za pomocą kalibrowanej probówki, umieścić płytkę obróconą o 90° w komorze (6.4.6) i prowadzić rozdział chromatograficzny w drugim kierunku (również w ciemności) do chwili, kiedy czoło rozpuszczalnika osiągnie linię narysowaną na powierzchni absorbentu. Wyjąć płytkę z komory i odparować rozpuszczalnik wymywający na powietrzu.
6.4.8. Umieścić płytkę w komorze chromatograficznej z parami jodu (6.3) na 10 minut i interpretować dwukierunkowy chromatogram, korzystając z wartości Rf i barw substancji odniesienia, rozdzielanych chromatograficznie w tym samym czasie (w tabeli II podano przewodnik po wartościach Rf i barwach).
Uwaga: W celu maksymalnego wybarwienia plam chromatogram należy pozostawić wystawiony na działanie powietrza na pół godziny po rozwinięciu.
6.4.9. Obecność barwników utleniających stwierdzoną zgodnie z pkt 6.4.8 można ostatecznie potwierdzić przez powtórzenie czynności opisanych w pkt 6.4.1–6.4.8 oraz przez dodanie w punkcie bazowym 1 do ilości ekstraktu podanej w pkt 6.4.2 1 µl substancji odniesienia zidentyfikowanej zgodnie z pkt 6.4.8. Jeżeli nie zostanie znaleziona dodatkowa plama w porównaniu z chromatogramem otrzymanym według pkt 6.4.8, interpretacja chromatogramu otrzymanego według pkt 6.4.8 jest prawidłowa.
TABELA II
Barwa substancji odniesienia po procesie chromatografii i wywołaniu parami jodu
| Substancja odniesienia | Barwa po wywołaniu parami jodu |
| R | beżowa |
| P | brązowa |
| alfa-N | fioletowa |
| beta-N | bladobrązowa |
| H | fioletowo-brązowa |
| MPD | żółtawobrązowa |
| PPD | fioletowo-brązowa |
| MTD | ciemnobrązowa |
| PTD | żółtawobrązowa |
| DAP | ciemnobrązowa |
| OAP | pomarańczowa |
| MAP | żółtawobrązowa |
| PAP | fioletowo-brązowa |
| 2-NPPD | brązowa |
| 4-NOPD | pomarańczowa |
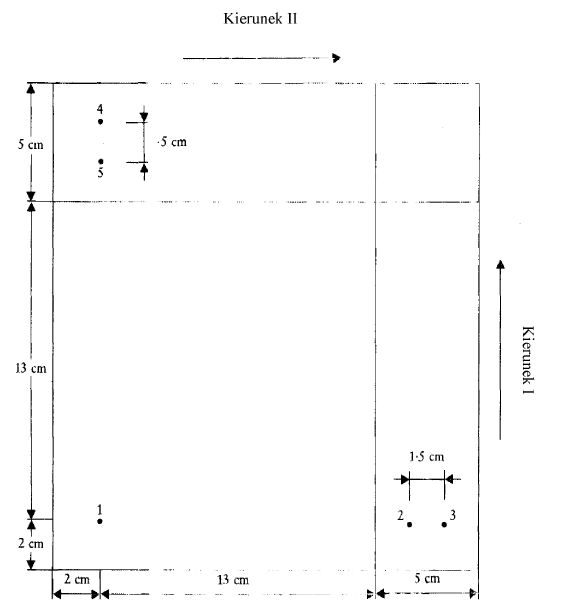
Rys. 6
X. IDENTYFIKACJA I OZNACZANIE AZOTYNÓW
A. IDENTYFIKACJA
1. CEL I ZAKRES
Metoda jest odpowiednia do identyfikowania azotynu w produktach kosmetycznych, szczególnie w kremach i pastach.
2. ZASADA
Na obecność azotynu wskazuje powstawanie barwnych pochodnych z fenylohydrazonem aldehydu 2-aminobenzoesowego (Nitrin®).
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. Rozcieńczony kwas siarkowy: rozcieńczyć 2 ml stężonego kwasu siarkowego (d 
3.2. Rozcieńczony kwas chlorowodorowy: rozcieńczyć 1 ml stężonego kwasu chlorowodorowego (d 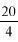
3.3. Metanol.
3.4. Roztwór fenylohydrazonu aldehydu 2-aminobenzoesowego (odczynnik Nitrin®) w metanolu.
Odważyć 2,0 g Nitrinu® i przenieść ilościowo do 100 ml kolby miarowej. Dodać kroplami 4 ml rozcieńczonego kwasu chlorowodorowego (3.2) i wstrząsnąć. Uzupełnić do kreski metanolem i mieszać do chwili, kiedy roztwór stanie się całkowicie przeźroczysty. Przechowywać roztwór w brązowej szklanej butli (4.3).
4. APARATURA
4.1. Zlewki, 50 ml.
4.2. Kolba miarowa, 100 ml.
4.3. Butla z brązowego szkła, 125 ml.
4.4. Płytka szklana, 10 x 10 cm.
4.5. Łopatka z tworzywa sztucznego.
4.6. Bibuła filtracyjna, 10 x 10 cm.
5. PROCEDURA
5.1. Rozsmarować część badanej próbki równomiernie na szklanej płytce (4.4), aby pokryć powierzchnię do grubości nie większej niż 1 cm.
5.2. Zanurzyć arkusz bibuły filtracyjnej (4.6) w wodzie destylowanej. Położyć bibułę na próbce i przycisnąć łopatką z tworzywa sztucznego (4.5).
5.3. Odczekać ok. jednej minuty i nanieść na środek bibuły filtracyjnej dwie krople rozcieńczonego kwasu siarkowego (3.1), a następnie dwie krople roztworu Nitrinu® (3.4).
5.4. Po 5–10 sekundach zdjąć bibułę filtracyjną i obejrzeć w świetle dziennym. Czerwonawo-purpurowe zabarwienie wskazuje na obecność azotynu.
Jeżeli zawartość azotynu jest niewielka, czerwonawo-purpurowa barwa zmienia się w żółtą po upływie od 5 do 15 sekund. Jeżeli zawartość azotynu jest niska, zmiana barwy z czerwonawo-purpurowej na żółtą następuje po upływie 15 sekund. W przypadku dużej zawartości azotynów zmiana następuje tylko po upływie jednej do dwóch minut.
6. UWAGA
Intensywność czerwonawo-purpurowej barwy i czas, który upływa przed jej zmianą na żółtą barwę, może być wskaźnikiem zawartości azotynu w próbce.
B. OZNACZANIE
1. CEL
Metoda opisuje oznaczanie azotynów w produktach kosmetycznych.
2. DEFINICJA
Zawartość azotynów w próbce oznaczona zgodnie z niniejszą metodą jest wyrażona w % masowych azotynu sodu.
3. ZASADA
Po rozcieńczeniu próbki wodą i wyklarowaniu roztworu azotan obecny w próbce zostaje poddany reakcji z sulfaniloamidem i N-1-naftyloetylenodwuaminą, a gęstość optyczną otrzymanej barwy oznacza się przy 538 nm.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy. 4.1. Odczynniki klarujące: odczynniki te nie mogą być używane dłużej niż przez tydzień od momentu ich przygotowania.
4.1.1. Odczynnik I Carreza
Rozpuścić 106 g żelazocyjanku (II) potasowego K4Fe(CN)6 3H2O w wodzie destylowanej i rozcieńczyć wodą do 1000 ml.
4.1.2. Odczynnik II Carreza
Rozpuścić 219,5 g octanu cynku Zn(CH3COO)2 2H20 i 30 ml lodowatego kwasu octowego w wodzie destylowanej i rozcieńczyć wodą do 1000 ml.
4.2. Roztwór azotynu sodu
Rozpuścić 0,500 g azotynu sodu w wodzie destylowanej w kolbie miarowej 1000 ml i rozcieńczyć wodą do kreski, rozcieńczyć 10,0 ml tego bazowego roztworu standardowego do 500 ml, 1 ml tego roztworu = 10 mikrogramów NaNO2.
4.3. 1 N roztwór wodorotlenku sodu.
4.4. 0,2% chlorowodorku sulfaniloamidu: rozpuścić 2,0 g sulfaniloamidu w 800 ml wody przez ogrzewanie. Schłodzić i dodać 100 ml stężonego kwasu chlorowodorowego, równocześnie mieszając. Rozcieńczyć wodą do 1000 ml.
4.5. 5 N kwas chlorowodorowy.
4.6. Odczynnik N-1-naftyl
Ten roztwór musi zostać przygotowany w dniu zastosowania. Rozpuścić 0,1 g dwuchlorowodorku N-1-naftyloetylenodwuaminy w wodzie i rozcieńczyć wodą do 100 ml.
5. APARATURA
5.1. Waga analityczna.
5.2. Kolby miarowe, 100, 250, 500 i 1000 ml.
5.3. Pipety ze zbiornikiem lub kalibrowane.
5.4. Cylindry miarowe, 100 ml.
5.5. Sączki filtracyjne karbowane o średnicy 15 cm, niezawierające azotynów.
5.6. Łaźnia wodna.
5.7. Spektrofotometr z naczynkami optycznymi o długości drogi optycznej 1 cm.
5.8. Pehametr.
5.9. Mikrobiureta, 10 ml.
5.10. Zlewki, 250 ml.
6. PROCEDURA
6.1. Odważyć ok. 0,5 g (m gramów) homogenizowanej próbki, z dokładnością do 0,1 mg, przenieść za pomocą gorącej wody destylowanej ilościowo do 250 ml zlewki (5.10) i uzupełnić objętość gorącą wodą destylowaną do ok. 150 ml. Umieścić zlewkę (5.10) w łaźni wodnej (5.6) o temperaturze 80°C na pół godziny. Od czasu do czasu wstrząsnąć zawartość.
6.2. Schłodzić do temperatury pokojowej i dodawać kolejno, równocześnie mieszając, 2 ml odczynnika I Carreza (4.1.1) i 2 ml odczynnika II Carreza (4.1.2).
6.3. Dodawać 1 N roztwór wodorotlenku sodu (4.3) aż do uzyskania pH 8,3 (używać pehametru (5.8)). Przenieść zawartość ilościowo do 250 ml kolby miarowej (5.2) i uzupełnić do kreski wodą destylowaną.
6.4. Wymieszać zawartość i przesączyć przez sączek karbowany (5.5).
6.5. Przenieść pipetą (5.3) odpowiednią podwielokrotność (V ml) klarownego filtratu, lecz nie więcej niż 25 ml, do 100 ml kolby miarowej (5.2) i dodać wodę destylowaną do objętości 60 ml.
6.6. Po zmieszaniu dodać 10,0 ml roztworu chlorowodorku sulfaniloamidu (4.4), a następnie 6,0 ml 5 N roztworu kwasu chlorowodorowego (4.5). Wymieszać i pozostawić do odstania na pięć minut. Dodać 2 ml odczynnika N-1 naftylowego (4.6), wymieszać i pozostawić do odstania na trzy minuty. Rozcieńczyć wodą do kreski i wymieszać.
6.7. Przygotować ślepą próbę, powtarzając czynności opisane w pkt 6.5 i 6.6, bez dodawania odczynnika N-1-naftylowego (4.6).
6.8. Zmierzyć za pomocą spektrofotometru (5.7) gęstość optyczną przy 538 nm roztworu otrzymanego według pkt 6.6, z zastosowaniem roztworu do ślepej próby (6.7) jako odniesienia.
6.9. Odczytać z wykresu kalibracyjnego (6.10) zawartość azotynu sodu w mikrogramach na 100 ml roztworu (m1 mikrogramów), która odpowiada gęstości optycznej zmierzonej, zgodnie z pkt 6.8.
6.10. Przygotować wykres kalibracyjny dla stężeń 0, 20, 40, 60, 80, 100 µg azotynu sodu w 100 ml, stosując roztwór azotynu sodu o stężeniu 10 µl na ml (4.2).
7. OBLICZENIE
Obliczyć zawartość azotynu sodu w procentach masowych za pomocą następującego wzoru:
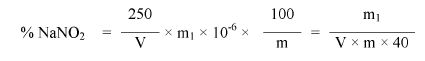
gdzie:
m = masa próbki pobranej do analizy w gramach (6.1),
m1 = zawartość azotynu sodu w mikrogramach oznaczona według pkt 6.9,
V = ilość mililitrów filtratu użytego do pomiaru (6.5).
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości ok. 0,2% m/m azotynu sodu różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć wartości absolutnej 0,005%.
XI. IDENTYFIKACJA I OZNACZANIE WOLNEGO FORMALDEHYDU
1. CEL I ZAKRES
Niniejsza metoda opisuje oznaczanie wolnego formaldehydu. Metoda może służyć do badania wszystkich produktów kosmetycznych i składa się z trzech części.
1.1. Identyfikacja.
1.2. Ogólne oznaczanie metodą kolorometryczną z pentano-2,4-dionem
Niniejsza metoda ma zastosowanie, gdy formaldehyd jest używany w czystej postaci lub z innymi konserwantami, które nie są donorami formaldehydu. W przeciwnym przypadku oraz gdy wyniki wykazują przekroczenie maksymalnego dozwolonego stężenia, musi zostać zastosowana następująca metoda potwierdzająca obecność.
1.3. Oznaczanie w obecności donorów formaldehydu
W metodzie opisanej powyżej (1.2) podczas derywatyzacji donory formaldehydu dzielą się, co prowadzi do zawyżania wyników (formaldehyd związany i spolimeryzowany).
Niezbędne jest oddzielenie wolnego formaldehydu za pomocą chromatografii cieczowej.
2. DEFINICJA
Zawartość wolnego formaldehydu w próbce oznaczona według tej metody jest wyrażona w procentach masowych.
3. IDENTYFIKACJA
3.1. Zasada
Wolny oraz związany formaldehyd w środowisku kwasu siarkowego nadaje odczynnikowi Schiffa barwę różową lub fioletowo-różową.
3.2. Odczynniki
Wszystkie odczynniki powinny być czyste do analizy, woda musi być demineralizowana.
3.2.1. Fuksyna.
3.2.2. Siarczyn sodu uwodniony 7 H2O.
3.2.3. Stężony kwas solny (d = 1,19).
3.2.4. Kwas siarkowy, ok. 1 M.
3.2.5. Odczynnik Schiffa
Należy odważyć do zlewki 100 mg fuksyny (3.2.1), po czym rozpuścić w 75 ml wody przy temperaturze 80°C. Po ostudzeniu należy dodać 2,5 g siarczynu sodu (3.2.2). Dopełnić do 100 ml. Odczynnik należy wykorzystać w ciągu dwóch tygodni.
3.3. Procedura
3.3.1. Odważyć 2 g próbki do 10 ml zlewki.
3.3.2. Dodać dwie krople kwasu siarkowego (3.2.4) oraz 2 ml odczynnika Schiffa (3.2.5). Przed próbą odczynnik Schiffa musi być bezwzględnie bezbarwny. Należy wstrząsnąć i pozostawić na pięć minut.
3.3.3. Jeżeli w ciągu pięciu minut będzie można zaobserwować różowy lub fioletowo-różowy odcień, formaldehyd jest obecny w nadmiarze ponad 0,01%, który musi zostać oznaczony metodą wolną i wiązaną (4), i jeżeli to konieczne musi zostać poddany procedurze (5).
4. OGÓLNE OZNACZANIE METODĄ KOLOROMETRYCZNĄ Z PENTANO-2,4-DIONEM
4.1. Zasada
Formaldehyd reaguje z pentano-2,4-dionem w obecności octanu amonu i tworzy 3,5-diacetylo-1,4-dihydrolutydynę. Za pomocą butan-1-olu dokonuje się ekstrakcji, po czym mierzy gęstość optyczną ekstraktu przy 410 nm.
4.2. Odczynniki
Wszystkie odczynniki muszą być czyste do analizy, woda musi być demineralizowana.
4.2.1. Bezwodny octan amonowy.
4.2.2. Stężony kwas octowy, d 
4.2.3. Pentan-2,4-dion świeżo destylowany pod zredukowanym ciśnieniem 25 mm Hg w temperaturze 25°C – nie powinien wykazywać absorpcji w 410 nm.
4.2.4. Butan-1-ol.
4.2.5. Kwas solny, 1 M.
4.2.6. Kwas solny, ok. 0,1 M.
4.2.7. Wodorotlenek sodu, 1 M.
4.2.8. Świeżo przygotowany roztwór skrobi zgodnie z Farmakopeą Europejską (1 g/50 ml wody), druga edycja 1980.
4.2.9. Formaldehyd o stężeniu masowym od 37 do 40%.
4.2.10. Standardowy roztwór jodu, 0,05 M.
4.2.11. Standardowy roztwór tiosiarczanu sodu, 0,1 M.
4.2.12. Odczynnik pentan 2,4-dion
W 1000 mm kolbie miarowej należy rozpuścić:
1) 150 g octanu amonu (4.2.1);
2) 2 ml pentan-2,4-dionu (4.2.3);
3) 3 ml kwasu octowego (4.2.2).
Dopełnić wodą do 1000 ml (pH roztworu ok. 6,4).
Odczynnik musi być świeżo przygotowany.
4.2.13. Odczynnik (4.2.12) bez pentan-2,4-dionu.
4.2.14. Standardowy formaldehyd: roztwór podstawowy
Należy wlać 5 g formaldehydu (4.2.9) do 1000 ml kolby miarowej i dopełnić wodą do 1000 ml.
Stężenie roztworu należy oznaczyć w następujący sposób: odlać 10 ml, dodać 25 ml standardowego roztworu jodu (4.2.10) oraz 10 ml roztworu wodorotlenku sodu (4.2.7), po czym odstawić na pięć minut.
Następnie należy zakwasić 11,00 ml kwasu solnego (4.2.5) i oznaczyć nadmiar jodu za pomocą standardowego roztworu tiosiarczanu sodu (4.2.11), używając jako wskaźnika roztworu skrobi (4.2.8).
1 ml z 0,05 M zużytego jodu (4.2.10) jest równy 1,5 mg formaldehydu.
4.2.15. Standardowy formaldehyd: roztwór rozcieńczony
Należy rozcieńczyć stopniowo wodą podstawowy roztwór formaldehydu w proporcjach 1/20, a potem 1/100.
1 ml tego roztworu zawiera ok. 1 µg formaldehydu.
Obliczyć dokładną zawartość.
4.3. Aparatura
4.3.1. Standardowe wyposażenie laboratoryjne.
4.3.2. Sączek do rozdziału faz, bibuła Whatman 1 PS lub równoważna.
4.3.3. Wirówka.
4.3.4. Kąpiel wodna ustawiona na temperaturę 60°C.
4.3.5. Spektrofotometr.
4.3.6. Naczynka szklane o długości ścieżki optycznej 1 cm.
4.4. Procedura.
4.4.1. Roztwór próbki
W 100 ml kolbie miarowej zważyć z dokładnością do 0,001 g ilość (w g) próbki analitycznej odpowiadającą przypuszczalnej zawartości formaldehydu, czyli ok. 150 µg.
Uzupełnić wodą do 100 ml i wymieszać (roztwór S).
Sprawdzić, czy pH jest zbliżone do 6; jeżeli nie, należy rozcieńczyć roztworem kwasu solnego (4.2.6).
Do 50 ml kolby stożkowej Erlenmeyera dodać:
1) 10,00 ml roztworu S;
2) 5,00 ml odczynnika pentano-2,4-dionu (4.2.12);
3) wodę demineralizowaną do końcowej objętości 30 ml.
4.4.2. Roztwór odniesienia
Możliwe zaburzenia wyników związane z podstawowym kolorem próbki analitycznej należy wyeliminować przez zastosowanie następującego roztworu odniesienia:
Do 50 ml kolby stożkowej należy wlać:
1) 10,00 ml roztworu S;
2) 5,00 ml odczynnika (4.2.13);
3) demineralizowaną wodę do końcowej objętości 30 ml.
4.4.3. Próba ślepa
Do 50 ml kolby stożkowej Erlenmeyera dodać:
1) 5,00 ml odczynnika pentano-2,4-dionu (4.2.12);
2) wodę demineralizowaną do końcowej objętości 30 ml.
4.4.4. Oznaczanie
4.4.4.1. Wstrząsać mieszaniny otrzymane zgodnie z pkt 4.4.1, 4.4.2 oraz 4.4.3 i zanurzyć na dokładnie 10 minut w łaźni wodnej, w temperaturze 60°C. Pozostawić do schłodzenia na dwie minuty w łaźni wodnej z lodem.
4.4.4.2. Przenieść mieszaniny do 50 ml rozdzielaczy zawierających po 10,00 ml butan-1-olu (4.2.4). Przepłukać każdą kolbę wodą w ilości od 3 do 5 ml, a następnie dolać tę wodę do rozdzielaczy. Potrząsać energicznie mieszaniną przez dokładnie 30 sekund. Pozostawić do rozdzielenia.
4.4.4.3. Należy oddzielić fazę butan-1-olu do naczynek pomiarowych (4.3.2), używając filtru rozdzielania faz. Może również zostać zastosowane wirowanie (3000 gn przez pięć minut).
4.4.4.4. Zmierzyć gęstość optyczną A1 ekstraktu roztworu próbki otrzymanego zgodnie z pkt 4.4.1 przy 410 nm w porównaniu z ekstraktem roztworu odniesienia otrzymanego zgodnie z pkt 4.4.2.
4.4.4.5. Podobnie zmierzyć ekstrakt roztworu do ślepej próby otrzymanego zgodnie z pkt 4.4.3 w porównaniu z butan-1-olem (A2).
Uwaga: Wszystkie czynności muszą być wykonane w ciągu 25 minut od chwili umieszczenia kolb stożkowych Erlenmeyera w łaźni wodnej o temperaturze 60°C.
4.4.5. Krzywa kalibracyjna
4.4.5.1. W 50 ml kolbie stożkowej Erlenmeyera umieścić:
1) 5,00 ml rozcieńczonego standardowego roztworu (4.2.15);
2) 5,00 ml pentano-2,4-dionu (4.2.12);
3) uzupełnić wodą demineralizowaną do końcowej objętości 30 ml.
4.4.5.2. Dalej postępować według wskazówek przedstawionych w pkt 4.4.4, zmierzyć gęstość optyczną w porównaniu z butan-1-olem (4.2.4).
4.4.5.3. Powtórzyć operację z 10, 15, 20 i 25 ml rozcieńczonego roztworu standardowego (4.2.15).
4.4.5.4. Dla otrzymania wartości zerowej (odnoszącej się do barwy odczynników) postępować, jak opisano w pkt 4.4.4.5.
4.4.5.5. Wykreślić krzywą kalibracyjną po odjęciu wartości zerowej od każdej gęstości optycznej otrzymanej zgodnie z pkt 4.4.5.1 i 4.4.5.3. Prawo Beera obowiązuje do 30 µg formaldehydu.
4.5. Obliczenia
4.5.1. Należy odjąć wartość A2 od A1 i odczytać z krzywej kalibracyjnej ilość C w mikrogramach formaldehydu w roztworze próbki (4.4.1).
4.5.2. Należy obliczyć zawartość formaldehydu w próbce (% m/m) za pomocą następującego wzoru:

gdzie:
m = masa próbki analitycznej w g.
4.6. Powtarzalność (patrz: norma ISO 5725)
W przypadku zawartości formaldehydu w ilości 0,2% różnica w wynikach dwóch równoległych oznaczeń, wykonanych na tej samej próbce, w metodzie kolorymetrycznej z pentano-2,4-dionem nie może przekraczać 0,005%.
Jeżeli oznaczenie wolnego formaldehydu prowadzi do wyników wyższych niż maksymalne stężenia przedstawione w załączniku V do rozporządzenia Parlamentu Europejskiego i Rady (WE) nr 1223/2009 z dnia 30 listopada 2009 r. dotyczącego produktów kosmetycznych (wersja przekształcona) (Dz. Urz. UE L 342 z 22.12.2009, str. 59, z późn. zm.), tj.:
1) gdy stężenie wynosi między 0,05% a 0,2% w nieoznakowanym produkcie,
2) lub gdy stężenie przekroczy 0,2%, to bez względu na to, czy jest to produkt oznakowany, czy nie
należy zastosować procedurę opisaną w pkt 5 poniżej.
5. OZNACZANIE W OBECNOŚCI DONORÓW FORMALDEHYDU
5.1. Zasada
Oddzielny formaldehyd w reakcji z pentano-2,4-dionem przekształca się w żółtą pochodną lutydyny w reaktorze postkolumnowym; pochodną tę można wykryć za pomocą gęstości optycznej przy 420 nm.
5.2. Odczynniki
Wszystkie odczynniki powinny być czyste do analizy, a woda musi być demineralizowana.
5.2.1. Woda jakości HPLC lub woda porównywalnej jakości.
5.2.2. Bezwodny octan amonu.
5.2.3. Stężony kwas octowy.
5.2.4. Pentano-2,4-dion (przechowywany w temperaturze 4°C).
5.2.5. Bezwodny fosforan disodowy.
5.2.6. 85% kwas ortofosforowy (d = 1,7).
5.2.7. Metanol o jakości HPLC.
5.2.8. Dichlorometan.
5.2.9. Formaldehyd o stężeniu masowym od 37 do 40%.
5.2.10. Wodorotlenek sodu, 1 M.
5.2.11. Kwas solny, 1 M.
5.2.12. Kwas solny, 0,002 M.
5.2.13. Świeżo przygotowany roztwór skrobi zgodnie z Farmakopeą Europejską (4.2.8).
5.2.14. Standardowy roztwór jodu, 0,05 M.
5.2.15. Standardowy roztwór tiosiarczanu sodu, 0,1 M.
5.2.16. Faza ruchoma
Wodny roztwór fosforanu disodu (5.2.5), 0,006 M o pH ustawionym za pomocą kwasu ortofosforowego (5.2.6) do 2,1.
5.2.17. Odczynniki postkolumnowe
W kolbie miarowej o objętości 1000 mm należy rozpuścić:
1) 62,5 g octanu amonu (5.2.2);
2) 7,5 ml kwasu octowego (5.2.3);
3) 5 ml pentano-2,4-dionu (5.2.4).
Należy dopełnić wodą do 1000 ml (5.2.1).
Ten odczynnik musi być chroniony przed światłem.
Czas zachowania: maksymalnie 3 dni w temperaturze 25°C.
Kolor nie powinien ulec zmianie.
5.2.18. Standardowy formaldehyd: roztwór podstawowy
Należy wlać 10 g formaldehydu (5.2.9) do kolby miarowej o objętości 1000 ml i dopełnić wodą do 1000 ml.
Należy oznaczyć stężenie roztworu w następujący sposób: odlać 5,00 ml, dodać 25,00 ml standardowego roztworu jodu (5.2.14) oraz 10,00 ml roztworu wodorotlenku sodu (5.2.10).
Odstawić na pięć minut.
Zakwasić 11,00 ml kwasu solnego (5.2.11), po czym miareczkować nadmiar standardowego roztworu sodu roztworem tiosiarczanu sodu (5.2.15) z użyciem roztworu skrobi (5.2.13) jako wskaźnika.
5.2.19. Standardowy formaldehyd: roztwór rozcieńczony
Należy rozcieńczyć roztwór podstawowy do jednej setnej jego pierwotnego stężenia w fazie ruchomej (5.2.16). 1 ml roztworu zawiera ok. 37 mg formaldehydu. Należy obliczyć dokładną zawartość.
5.3. Aparatura
5.3.1. Standardowe wyposażenie laboratoryjne.
5.3.2. Bezpulsacyjna pompa HPLC.
5.3.3. Niskociśnieniowa bezpulsacyjna pompa do odczynników (lub druga pompa HPLC).
5.3.4. Zawór wtryskowy z 10 µl pętlą.
5.3.5. Reaktor postkolumnowy powinien być wyposażony w:
1) jedną 1-litrową kolbę o trzech szyjkach;
2) jeden 1-litrowy ogrzewacz do kolb;
3) dwie kolumny Vigreux z przynajmniej 10 płytami, z których dwie powinny być chłodzone powietrzem;
4) nierdzewną rurę stalową (do odprowadzania ciepła) 1,6 mm o średnicy wewnętrznej 0,23 mm i długości 400 mm;
5) rurę teflonową 1,6 mm o średnicy wewnętrznej 0,30 mm, długości 5 m („francuskie szycie” – patrz: dodatek 1);
6) jeden trójnik bez martwej objętości (Valco lub odpowiednik);
7) trzy złączki bez żadnych martwych objętości;
Lub: jeden moduł post kolumnowy Applied Biosystems PCRS 520 lub odpowiednik wyposażony w 1 ml reaktor.
5.3.6. Filtr membranowy o rozmiarze porów 0,45 µm.
5.3.7. Zasobnik SEP-PAKR C18 lub odpowiednik.
5.3.8. Kolumny gotowe do użytku:
1) typu Bischoff hypersil RP 18 (typu NC odnośnik C 25.46 1805)
(5 µm, długość = 250 mm, średnica wewnętrzna = 4,6 mm) lub
2) Dupont, Zorbax ODS
(5 µm, długość = 250 mm, średnica wewnętrzna = 4,6 mm), lub
3) Phase SEP, spherisorb ODS 2
(5 µm, długość = 250 mm, średnica wewnętrzna = 4,6 mm).
5.3.9 Przedkolumna
typu Bischoff K1 hypersil RP 18 (odnośnik K1 G 6301 1805)
(5 µm, długość = 10 mm, lub jej odpowiednik).
5.3.10. Kolumna z kolumną wstępną są połączone za pomocą systemu Ecotube (odnośnik A 15 020 508 Bischoff) lub odpowiednika.
5.3.11. Należy złożyć aparaturę (5.3.5) tak, jak to przedstawiono na schemacie blokowym w dodatku 2.
Połączenia za zaworem wtryskowym muszą być możliwie krótkie. W tym przypadku nierdzewna stalowa rura między wyjściem reaktora a wejściem detektora będzie pełniła rolę chłodnicy mieszaniny przed detekcją, temperatura wewnątrz reaktora będzie nieznana, ale stała.
5.3.12. Detektor widzialnego promieniowania UV.
5.3.13. Rejestrator.
5.3.14. Wirówka.
5.3.15. Kąpiel z użyciem ultradźwięków.
5.3.16. Mieszadło wibracyjne (Vortex lub odpowiednik).
5.4. Procedura
5.4.1. Krzywa kalibracyjna
Tworzy się ją dzięki nanoszeniu wartości szczytowych jako funkcji stężenia rozcieńczonego formaldehydu do roztworu odniesienia formaldehydu. Należy przygotować roztwory standardowe przez rozcieńczenie roztworu odniesienia (5.2.19) fazą ruchomą (5.2.16):
1) 1,00 ml roztworu (5.2.19) rozpuszczonego w 20,00 ml (ok. 185 µg / 100 ml);
2) 2,00 ml roztworu (5.2.19) rozpuszczonego w 20,00 ml (ok. 370 µg/100 ml);
3) 5,00 ml roztworu (5.2.19) rozcieńczonego w 25,00 ml (ok. 740 µg / 100 ml);
4) 5,00 ml roztworu (5.2.19) rozcieńczonego w 20,00 ml (ok. 925 µg / 100 ml). Roztwory standardowe są przechowywane przez godzinę w temperaturze laboratoryjnej i muszą być świeżo przygotowane.
Liniowość krzywej kalibracyjnej jest prawidłowa dla stężeń między 1,00 a 15,00 µg/ml.
5.4.2. Przygotowanie próbek
5.4.2.1. Emulsje (kremy, podkłady do makijażu, kredki do oczu)
Do zamkniętej 100 ml kolby należy odważyć z dokładnością do 0,001 g ilość próbki analitycznej (mg) odpowiadającej przypuszczalnie ilości 100 µg formaldehydu. Następnie należy dodać 20,00 ml dichlorometanu (5.2.8) oraz 20,00 ml dokładnie odmierzonego kwasu solnego (5.2.12). Należy wymieszać składniki za pomocą mieszadła wibrującego (5.3.16) oraz kąpieli ultradźwiękowej (5.3.15), a następnie oddzielić dwie fazy przez wirowanie (3000 gn przez dwie minuty). W międzyczasie należy oczyścić zasobnik (5.3.7) 2 ml metanolu (5.2.7), po czym przystosować zasobnik 5 ml wody (5.2.1).
Przepuścić 4 ml fazy płynnej ekstraktu przez przystosowany zasobnik, odrzucić pierwsze 2 ml, a następnie odzyskać następującą po tym frakcję.
5.4.2.2. Balsamy, szampony
Do zamkniętej kolby o objętości 100 ml odważyć z dokładnością do 0,001 g ilość próbki analitycznej (mg) odpowiadającej przypuszczalnie ok. 500 µg formaldehydu. Dopełnić fazą ruchomą do 100 ml (5.2.16).
Przepuścić roztwór przez filtr (5.3.6) i wprowadzić lub przepuścić przez przystosowany jak poprzednio zasobnik (5.4.2.1). Wszystkie roztwory muszą zostać wprowadzone bezpośrednio po przygotowaniu.
5.4.3. Warunki chromatografii:
1) prędkość przepływu fazy ruchomej: 1 ml/min;
2) prędkość przepływu odczynników: 0,5 ml/min;
3) całkowita prędkość przepływu u wyjścia detektora: 1,5 ml/min;
4) wprowadzona objętość: 10 µl;
5) temperatura elucji: w przypadku utrudnionego oddzielania zanurzyć kolumnę w kąpieli z topniejącego lodu, poczekać od 15 do 20 min, zanim temperatura się ustabilizuje;
6) temperatura reakcji w postkolumnie: 100°C;
7) detekcja: przy 420 nm.
Uwaga: Cały system do chromatografii oraz postkolumna po użyciu muszą być przepłukane wodą (5.2.1). Jeżeli urządzenia są nieużywane przez więcej niż dwa dni, po przepłukaniu wodą należy przepłukać aparaturę metanolem (5.2.7). Przed ponownym przystosowaniem systemu, w celu uniknięcia rekrystalizacji, należy przepuścić przez system wodę.
5.5. Obliczenia
Emulsje: (5.4.2.1):
Zawartość formaldehydu w % (m/m) oblicza się według wzoru:
Balsamy, szampony:
W tym przypadku stosuje się wzór zmieniony w następujący sposób:
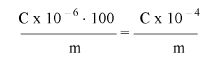
gdzie:
m = masa badanej próbki w g (5.4.2.1),
C = koncentrat formaldehydu w µg/100 ml odczytany z krzywej kalibracyjnej (5.4.1).
5.6. Powtarzalność (patrz: norma ISO 5725)
Dla 0,05% zawartości formaldehydu różnica między wynikami dwóch oznaczeń w równolegle przeprowadzonych pomiarach tej samej próbki nie powinna przekraczać 0,001%.
Dla 0,2% zawartości formaldehydu różnica między wynikami dwóch oznaczeń w równolegle przeprowadzonych pomiarach tej samej próbki nie powinna przekraczać 0,005%.
DODATEK 1
Instrukcja przygotowania przyrządu do „francuskiego szycia”
Wymagane akcesoria
Jedna drewniana szpula o średnicy zewnętrznej 5 cm z otworem o średnicy 1,5 cm przechodzącym przez środek. Wstawić 4 stalowe gwoździe (jak pokazano na rys. 7 i 8). Odległość między dwoma gwoździami musi wynosić 1,8 cm, muszą być one oddalone o 0,5 cm od otworu.
Jedna sztywna igła (typu hakowego) do tworzenia pętli rurki teflonowej.
Rurka teflonowa 1,6 mm o długości pięciu metrów i średnicy wewnętrznej 0,3 mm.
Procedura
Aby rozpocząć „francuskie szycie” za pomocą przyrządu, należy przeprowadzić rurkę teflonową od góry do dołu szpuli przez jej środkowy otwór (pozostawiając ok. 10 cm rurki odstające od spodu szpuli, aby umożliwić przeciągnięcie łańcucha w procesie tkania); następnie należy zawinąć rurkę kolejno o cztery gwoździe, jak pokazano na rys. 9.
Wejście i wyjście przyrządu jest ochraniane metalowymi zaciskami i śrubami zaciskowymi; należy uważać, aby nie uszkodzić teflonu przy silnym pociągnięciu.
Zawinąć rurkę wokół każdego gwoździa po raz drugi i wykonać „ściegi” w następujący sposób: za pomocą sztywnej igły unieść rurkę z niższego rzędu nad rurkę z górnego rzędu (rys. 10); czynność tę powtórzyć na każdym z gwoździ w kolejności (1, 2, 3, 4 w kierunku przeciwnym do ruchu wskazówek zegara) aż do uzyskania łańcuszka o długości 5 m lub do uzyskania pożądanej długości.
Należy pozostawić ok. 10 cm w celu zamknięcia łańcucha. W celu zakończenia łańcucha należy przeprowadzić rurkę przez każdą z czterech pętli i pociągnąć delikatnie.
Uwaga: Przyrządy do tkania łańcucha wytwarzane do zastosowania w reaktorach postkolumnowych są dostępne na rynku (Supelco).
Schematyczny wykres szpuli
Rys. 7
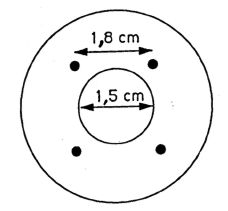
Rys. 8
Rys. 9
Pierwsza rurka
Rys. 10
Druga rurka
Aby utworzyć „ścieg”, należy unieść niższą rurkę (linia ciągła) nad drugą rurkę (linia przerywana)
Rys. 11
DODATEK 2
1 = pompa HPLC
2 = zawór wtryskowy
3 = kolumna z kolumną wstępną
4 = pompa do odczynników
5 = trójnik bez martwej objętości
5' = trójnik (Vortex)
6–6' = złączki bez martwych objętości
7 = przyrząd do „francuskiego szycia”
7' = reaktor
8 = kolba o trzech szyjkach
9 = ogrzewacz do kolb
10 = chłodziwo
11 = nierdzewna rura stalowa do wymiany ciepła
11' = wymiennik ciepła
12 = detektor widzialnego promieniowania UV
13 = moduł postkolumnowy PCRS 520
XII. OZNACZANIE REZORCYNY W SZAMPONACH I PŁYNACH DO WŁOSÓW
1. CEL I ZAKRES
Podaną metodą chromatografii gazowej oznacza się rezorcynę w szamponach i płynach do włosów. Metoda jest odpowiednia dla stężeń od 0,1 do 2,0% w masie próbki.
2. DEFINICJA
Zawartość rezorcyny w próbce określona tą metodą jest wyrażana w procentach masowych.
3. ZASADA
Rezorcynę i 3,5-dwuhydroksytoluen (5-metylorezorcyna), dodawane jako standard wewnętrzny, wydziela się z próbki metodą chromatografii cienkowarstwowej. Oba związki są izolowane przez zdrapywanie ich plam z płytki cienkowarstwowej i ekstrahowanie metanolem. Na zakończenie ekstrahowane związki suszy się, sililuje i oznacza metodą chromatografii gazowej.
4. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
4.1. Kwas chlorowodorowy, 25% (m/m).
4.2. Metanol.
4.3. Etanol, 96% (v/v).
4.4. Gotowe płytki do chromatografii cienkowarstwowej (TLC), pokryte żelem krzemionkowym, ze sztucznego tworzywa lub aluminiowe, ze wskaźnikiem fluorescencyjnym. Dezaktywację przeprowadza się w następujący sposób: spryskiwać wodą standardowe płytki, pokryte żelem krzemionkowym, do chwili, aż staną się szkliste. Pozostawić spryskane płytki do wyschnięcia w temperaturze pokojowej na jedną godzinę do trzech godzin.
Uwaga: Jeżeli płytki nie zostaną dezaktywowane, mogą wystąpić straty rezorcyny na skutek nieodwracalnej adsorpcji na żelu krzemionkowym.
4.5. Roztwór rozwijający: aceton/chloroform/kwas octowy (20:75:5 obj.).
4.6. Standardowy roztwór rezorcyny: rozpuścić 400 mg rezorcyny w 100 ml 96% etanolu (4.3) (1 ml odpowiada 4000 µg rezorcyny).
4.7. Roztwór standardu wewnętrznego: rozpuścić 400 mg 3,5-dwuhydroksytoluenu (DHT) w 100 ml 96% etanolu (4.3) (1 ml odpowiada 4000 µg DHT).
4.8. Mieszanina standardowa: zmieszać 10 ml roztworu 4.6 i 10 ml roztworu 4.7 w 100 ml kolbie miarowej, uzupełnić 96% etanolem (4.3) do kreski i wymieszać (1 ml odpowiada 400 µg rezorcyny i 400 µg DHT).
4.9. Środki sililujące.
4.9.1. N, O-bis-(trójmetylosililo)-trójfluoroacetamid (BSTFA).
4.9.2. Heksametylodwusilazan (HMDS).
4.9.3. Trójmetylochlorosilan (TMCS).
5. APARATURA
5.1. Zwykłe wyposażenie do chromatografii cienkowarstwowej i gazowej.
5.2. Szkło laboratoryjne.
6. PROCEDURA
6.1. Przygotowanie próbki
6.1.1. Zważyć dokładnie próbkę analityczną (m gramów) produktu do 150 ml zlewki, która zawiera ok. 20 do 50 mg rezorcyny.
6.1.2. Zakwaszać kwasem solnym (4.1) do momentu, kiedy mieszanina stanie się kwaśna (średnio potrzeba 2 do 4 ml), dodać 10 ml (40 mg DHT) roztworu standardowego wewnętrznego (4.7) i wymieszać. Przenieść do 100 ml kolby miarowej z etanolem (4.3), uzupełnić etanolem do kreski i wymieszać.
6.1.3. Nanieść 250 µl roztworu (6.1.2) na dezaktywowaną płytkę z żelem krzemionkowym (4.4), w formie ciągłej linii o długości ok. 8 cm. Należy zadbać o to, aby linia była możliwie jak najwęższa.
6.1.4. Nanieść 250 µl mieszaniny standardowej (4.8) na tę samą płytkę, w taki sam sposób jak opisano w pkt 6.1.3.
6.1.5. Nakropić w dwóch pkt linii początkowej po 5 µl każdego z roztworów (4.6 i 4.7) w celu łatwiejszego umiejscowienia po rozwinięciu płytki.
6.1.6. Rozwijać płytkę w komorze pozbawionej podkładu (nienasyconej), wypełnionej rozpuszczalnikiem rozwijającym (4.5), do momentu, w którym czoło rozpuszczalnika osiągnie linię w odległości 12 cm od linii początkowej; zwykle trwa to ok. 45 minut. Wysuszyć płytkę na powietrzu i umiejscowić strefę rezorcyny/DHT w świetle nadfioletowym o krótkich falach (254 nm). Oba związki mają w przybliżeniu te same wartości Rf. Oznaczyć pasma ołówkiem, w odległości 2 mm od zewnętrznej linii brzegowej pasm. Usunąć te strefy z płytki i zebrać adsorbent z każdego pasma do kolby 10 ml.
6.1.7. Ekstrahować adsorbent zawierający próbkę i adsorbent zawierający mieszaninę standardową w następujący sposób: dodać 2 ml metanolu (4.2) i ekstrahować przez godzinę, nieustannie mieszając. Przesączyć mieszaninę i powtórzyć ekstrakcję przez kolejne 15 minut z 2 ml metanolu.
6.1.8. Połączyć ekstrakty i odparować rozpuszczalnik, susząc przez noc w eksykatorze zawierającym odpowiedni środek suszący. Nie stosować ogrzewania.
6.1.9. Sililować pozostałości (6.1.8) stosownie do wskazówek podanych w pkt 6.1.9.1 lub 6.1.9.2.
6.1.9.1. Dodać 200 µl BSTFA (4.9.1) mikrostrzykawką i pozostawić mieszaninę w zamkniętym naczyniu na 12 godzin, w temperaturze pokojowej.
6.1.9.2. Dodać kolejno 200 µl HMDS (4.9.2) i 100 µl TMCS (4.9.3) mikrostrzykawką i ogrzewać mieszaninę przez 30 minut, w temperaturze 60°C, w zamkniętym naczyniu. Mieszaninę schłodzić.
6.2. Chromatografia gazowa
6.2.1. Warunki chromatograficzne
Kolumna musi mieć zdolność rozdzielczą R równą lub większą niż 1,5, gdzie:
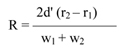
gdzie:
r1 i r2 = czas retencji w minutach dla dwóch pików,
w1 i w2 = szerokość tych dwóch pików w połowie wysokości w mm,
d' = szybkość przesuwu papieru w mm na minutę.
Następujące kolumny i warunki chromatografii gazowej uznano za odpowiednie:
| Kolumna | materiał: | stal nierdzewna |
|
| długość: | 200 cm |
|
| średnica wewnętrzna: | ~ 3 mm |
|
| wypełnienie: | 10% OV–17 na Chromosorbie WAW 100 do 120 mesh |
|
|
|
|
| Detektor płomieniowo-jonizacyjny | ||
| Temperatura: |
|
|
|
| kolumna: | 185°C (izotermiczna) |
|
| detektor: | 250°C |
|
| dozownik: | 250°C |
| Gaz nośny: | azot |
|
| Przepływ: | 45 ml/min. |
|
Dla ustawienia przepływów wodoru i powietrza należy przestrzegać instrukcji producenta.
6.2.2. Wprowadzić strzykawką od 1 do 3 µl roztworów otrzymanych w sposób opisany w pkt 6.1.9 do chromatografu gazowego. Wykonać pięć iniekcji dla każdego roztworu (6.1.9), zmierzyć powierzchnię pików, obliczyć średnią wartość i stosunek powierzchni pików: S = powierzchnia piku rezorcyny / powierzchnia piku DHT.
7. OBLICZENIE
Stężenie rezorcyny w próbce, w procentach masowych (% m/m), jest wyrażone za pomocą wzoru:
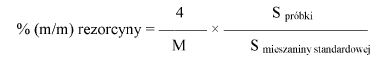
gdzie:
M = próbka analityczna w gramach (6.1.1),
S próbki = średni stosunek powierzchni pików według pkt 6.2.2 dla roztworu próbki,
S mieszaniny standardowej = średni stosunek powierzchni pików według pkt 6.2.2 dla mieszaniny standardowej.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości rezorcyny ok. 0,5% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć wartości absolutnej 0,025%.
XIII. OZNACZANIE METANOLU W STOSUNKU DO ETANOLU LUB PROPAN-2-OLU
1. CEL I ZAKRES
Metoda opisuje analizę metanolu metodą chromatografii gazowej we wszystkich produktach kosmetycznych (łącznie z aerozolowymi). Oznaczać można względne poziomy stężeń od 0 do 10%.
2. DEFINICJA
Zawartość metanolu oznaczana tą metodą jest wyrażana w procentach masowych metanolu w stosunku do etanolu lub propan-2-olu.
3. ZASADA
Oznaczanie wykonuje się metodą chromatografii gazowej.
4. ODCZYNNIKI
Należy używać odczynników czystych do analizy.
4.1. Metanol.
4.2. Absolutny etanol.
4.3. Propan-2-ol.
4.4. Chloroform wolny od alkoholi przez przemycie wodą.
5. APARATURA
5.1. Chromatograf gazowy
z detektorem katarometrycznym do próbek aerozoli
z detektorem płomieniowo-jonizacyjnym do próbek nieaerozolowych.
5.2. Kolby miarowe, 100 ml.
5.3. Pipety, 2 ml, 20 ml, 0–1 ml.
5.4. Mikrostrzykawki od 0 do 100 µl i od 0 do 5 µl oraz (tylko do próbek aerozolowych) specjalne strzykawki gazoszczelne z zaworem suwakowym (patrz: rys. 5 w części II „Praktyczne przygotowanie próbek laboratoryjnych”).
6. PROCEDURA
6.1. Przygotowanie próbek
6.1.1. Próbki wyrobów aerozolowych przygotowuje się zgodnie z przepisami części II „Praktyczne przygotowanie próbek laboratoryjnych”, a następnie poddaje się je analizie metodą chromatografii gazowej w warunkach przedstawionych w pkt 6.2.1.
6.1.2. Próbki wyrobów nieaerozolowych, przygotowane zgodnie z przepisami wymienionej powyżej części II, rozcieńcza się wodą do poziomu stężenia od 1 do 2% etanolu lub propan-2-olu, a następnie poddaje się je analizie metodą chromatografii gazowej w warunkach przedstawionych w pkt 6.2.2.
6.2. Chromatografia gazowa.
6.2.1. Dla próbek aerozolowych używa się detektora katarometrycznego.
6.2.1.1. Wypełnienie kolumny: 10% Hallcomid M 18 na Chromosorbie WAW, od 100 do 200 mesh.
6.2.1.2. Kolumna musi mieć zdolność rozdzielczą R równą lub większą niż 1,5, obliczoną według wzoru:
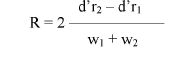
gdzie:
r1 i r2 = czas retencji w minutach dla dwóch pików,
w1 i w2 = szerokość tych dwóch pików w połowie wysokości,
d' = szybkość przesuwu papieru w mm na minutę.
6.2.1.3. Na uzyskanie tej rozdzielczości pozwalają następujące warunki:
| Kolumna | materiał: | stal nierdzewna |
|
| długość: | od 3 do 5 metrów |
|
| średnica: | 3 mm |
| Natężenie prądu mostka katarometru: | 150 mA | |
| Gaz nośny: |
| hel |
|
| ciśnienie: | 2,5 bar |
|
| przepływ: | 45 ml/min |
| Temperatura: |
|
|
|
| dozownik: | 150°C |
|
| detektor: | 150°C |
|
| piec kolumny: | 65°C |
Dokładność pomiarów powierzchni pików można poprawić przez zastosowanie całkowania elektronicznego.
6.2.2. Dla próbek nieaerozolowych:
6.2.2.1. Kolumnę wypełnia się: Chromosorb 105 lub Porapak QS oraz stosuje się detektor płomieniowo-jonizacyjny.
6.2.2.2. Kolumna musi mieć zdolność rozdzielczą R równą lub większą niż 1,5, obliczoną według wzoru:
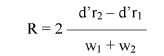
gdzie:
r1 i r2 = czas retencji w minutach dla dwóch pików,
w1 i w2 = szerokości tych dwóch pików w połowie wysokości w mm,
d' = szybkość przesuwu papieru w mm na minutę.
6.2.2.3. Tę rozdzielczość uzyskano w następujących warunkach:
| Kolumna | materiał: | stal nierdzewna |
|
| długość: | 2 metry |
|
| średnica: | 3 mm |
| Czułość elektrometru: | 8 x 10-10 A | |
| Gazy: |
|
|
|
| nośnik: | azot |
|
| ciśnienie: | 2,1 bar |
|
| przepływ: | 40 ml/min |
| Gaz pomocniczy: | wodór | |
|
| ciśnienie: | 1,5 bar |
|
| przepływ: | 20 ml/min |
| Temperatura: |
|
|
|
| dozownik: | 150°C |
|
| detektor: | 230°C |
|
| piec kolumny: | od 120 do 130°C |
7. WYKRES STANDARDOWY
7.1. W celu wykonania analizy metodą chromatografii gazowej według pkt 6.2.1 (kolumna Hallcomid M 18) należy stosować poniższe mieszaniny standardowe. Mieszaniny przygotowuje się przez odmierzenie pipetami, lecz należy ustalić dokładną ilość składnika przez bezpośrednie zważenie pipety lub kolby po każdym dodaniu składnika.
| Stężenie względne (m/m%) | Metanol (ml) | Etanol lub propan-2-ol (ml) | Chloroform dodany do objętości (ml) |
| w przybliżeniu 2,5% | 0,5 | 20 | 100 ml |
| w przybliżeniu 5,0% | 1,0 | 20 | 100 ml |
| w przybliżeniu 7,5% | 1,5 | 20 | 100 ml |
| w przybliżeniu 10,0% | 2,0 | 20 | 100 ml |
Wprowadzić od 2 do 3 µl do chromatografu, zapewniając warunki podane w pkt 6.2.1.
Obliczyć stosunek powierzchni pików (metanol/etanol) lub (metanol/propan-2-ol) dla wszystkich mieszanin.
Wykreślić standardowy wykres z użyciem oznaczeń:
oś X: % metanolu w stosunku do etanolu lub propan-2-olu,
oś Y: stosunek powierzchni pików (metanol / etanol) lub (metanol / propan-2-ol).
7.2. W celu wykonania analizy metodą chromatografii gazowej według pkt 6.2.2 (kolumna Porapak QS lub Chromosorb 105) należy stosować poniższe mieszaniny standardowe. Mieszaniny przygotowuje się przez odmierzanie mikrostrzykawką i pipetą, lecz należy ustalić dokładną ilość składnika przez bezpośrednie zważenie pipety lub kolby po każdym dodaniu składnika.
7.3. Standardowy wykres musi być linią prostą.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości metanolu 5% w stosunku do etanolu lub propan-2-olu różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,25%.
XIV. OZNACZANIE DICHLOROMETANU i 1,1,1-TRICHLOROETANU
1. CEL I ZAKRES STOSOWANIA
Przedstawiona metoda obejmuje oznaczanie dichlorometanu (chlorek metylenu) i 1,1,1-trichloroetanu (chloroform metylowy) we wszystkich produktach kosmetycznych mogących zawierać te rozpuszczalniki.
2. DEFINICJA
Zawartość dichlorometanu i 1,1,1-trichloroetanu w próbce wyrobu oznaczana zgodnie z tą metodą jest wyrażona w procentach masowych.
3. ZASADA
W metodzie jest używana chromatografia gazowa z chloroformem jako wewnętrznym standardem.
4. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
4.1. Chloroform (CHCl3).
4.2. Czterochlorek węgla (CCl4).
4.3. Dichlorometan (CH2Cl2).
4.4. 1,1,1-trichloroetan (CH3CCl3).
4.5. Aceton.
4.6. Azot.
5. APARATURA
5.1. Zwykły sprzęt laboratoryjny.
5.2. Chromatograf gazowy wyposażony w detektor przewodnictwa cieplnego.
5.3. Butelka do przenoszenia, od 50 do 100 ml (patrz: pkt 5.3 w części II „Praktyczne przygotowanie próbek laboratoryjnych”).
5.4. Ciśnieniowa strzykawka gazowa, 25 lub 50 µl (patrz: pkt 5.4.2.2 w części II „Praktyczne przygotowanie próbek laboratoryjnych”).
6. PROCEDURA
6.1. Próbka pod normalnym ciśnieniem: odważyć dokładnie próbkę w kolbie stożkowej zamykanej korkiem. Wprowadzić dokładnie zważoną ilość chloroformu (4.1) jako standard wewnętrzny równoważny przewidywanej ilości dichlorometanu i 1,1,1-trichloroetanu zawartych w próbce. Dokładnie wymieszać.
6.2. Próbka pod zwiększonym ciśnieniem: zastosować metodę pobierania próbek opisaną w części II „Praktyczne przygotowanie próbek laboratoryjnych”, ale z następującymi poprawkami:
6.2.1. Po przeniesieniu próbki do butelki do przenoszenia (5.3) wprowadzić następnie do tej butelki pewną objętość chloroformu (4.1) jako standard wewnętrzny, równoważny przewidywanej ilości dichlorometanu i/lub 1,1,1-trichloroetanu zawartych w próbce. Dokładnie wymieszać. Przemyć martwą objętość zaworu 0,5 ml czterochlorku węgla (4.2). Po wysuszeniu oznaczyć różnicowo dokładnie dodaną masę standardu wewnętrznego.
6.2.2. Po napełnieniu strzykawki próbką dysza strzykawki powinna być przepłukana azotem (4.6), tak aby żadna pozostałość próbki nie została w niej przed wprowadzeniem do chromatografu.
6.2.3. Po każdym pobraniu próbki powierzchnie zaworu i części przenoszącej powinny być przepłukane kilkakrotnie acetonem (4.5) (z użyciem strzykawki do iniekcji podskórnych) i wysuszone dokładnie azotem (4.6).
6.2.4. Dla każdej analizy należy wykonać pomiary z użyciem dwóch różnych butelek do przenoszenia i powtarzać pięciokrotnie pomiary dla każdej butelki.
7. WARUNKI ANALIZY CHROMATOGRAFICZNEJ
7.1. Kolumna wstępna:
Połączenie rurowe: stal nierdzewna.
Długość: 300 mm.
Średnica: 3 lub 6 mm.
Wypełnienie: to samo, które jest używane do wypełnienia kolumny analitycznej.
7.2. Kolumna
Faza stacjonarna jest przygotowana z Hallcomidu M l8 osadzonego na Chromosorbie. Kolumna powinna mieć zdolność rozdzielczą „R” równą lub wyższą niż 1,5, obliczoną według wzoru:
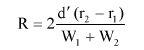
gdzie:
r1 i r2 = czas retencji (w minutach),
W1 i W2 = szerokości piku w połowie wysokości (w milimetrach),
d' = szybkość przesuwu papieru (w milimetrach na minutę).
7.3. W przykładzie poniżej podano, jakie otrzymano wyniki w zależności od zastosowanej kolumny
| Kolumna | I | II |
| Materiał: | Rurka ze stali nierdzewnej | Rurka ze stali |
| Długość: | 350 cm | 400 cm |
| Średnica: | 3 mm | 6 mm |
| Nośnik: |
|
|
| Chromosorb: | WAW | WAW-DMCS-HP |
| analiza sitowa: | 100–120 oczek | 60–80 oczek |
| Faza stacjonarna: | Hallcomid M 18, 10% | Hallcomid M 18, 20% |
Warunki temperaturowe mogą się różnić zależnie od zastosowanego aparatu. W podanych przykładach ustalono je następująco:
| Kolumna |
| I | II |
| Temperatura: |
|
|
|
|
| kolumna: | 65°C | 75°C |
|
| dozownik: | 150°C | 125°C |
|
| detektor: | 150°C | 200°C |
| Gaz nośny: |
|
|
|
|
| szybkość przepływu helu: | 45 ml/min | 60 ml/min |
|
| ciśnienie wlotowe: | 2,5 bara | 2 bary |
| Dozowanie: |
| 15 µl | 15 µl |
8. MIESZANINA DO USTALENIA WSPÓŁCZYNNIKÓW REAKCJI
Sporządzić następującą dokładnie odważoną mieszaninę w kolbie stożkowej z korkiem: dichlorometan (4.3), 30% (m/m), 1,1,1-trichloroetan (4.4), 35% (m/m), chloroform (4.1), 35% (m/m).
9. OBLICZENIA
9.1. Obliczyć współczynnik reakcji substancji „p” w stosunku do substancji „a” wybranej jako standard wewnętrzny.
Podać najpierw dane o pierwszej substancji oznaczonej „p”, gdzie:
kp = jej współczynnik reakcji,
mp = jej masa w mieszaninie,
Ap = powierzchnia jej piku.
Następnie podać dane o substancji „a”, przyjmując:
ka = jej współczynnik reakcji,
Ma = jej masa w mieszaninie,
Aa = powierzchnia jej piku.
Według wzoru:
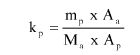
Przykładowo otrzymano następujące współczynniki reakcji (przyjmując dla chloroformu k = 1):
dichlorometan: k1 = 0,78 +/– 0,03
1,1,1-trichloroetan: k2 = 1,00 +/– 0,03
9.2. Obliczyć obecność w % (m/m) dichlorometanu i 1,1,1-trichloroetanu w analizowanej próbce, przyjmując:
ma = masa (w gramach) wprowadzonego chloroformu,
Ms = masa (w gramach) analizowanej próbki,
Aa = powierzchnia piku chloroformu,
A1 = powierzchnia piku dichlorometanu,
A2 = powierzchnia piku 1,1,1-trichloroetanu,
według wzoru:
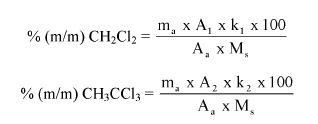
10. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości dichlorometanu i/lub 1,1,1-trichloroetanu 25% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać wartości 2,5% (m/m).
XV. IDENTYFIKOWANIE I OZNACZANIE CHINOLIN-8-OLU I SIARCZANU BIS(8-HYDROKSYCHINOLINY)
1. CEL I ZAKRES STOSOWANIA
Metoda opisuje identyfikowanie i ilościowe oznaczanie chinolin-8-olu i siarczanu bis(8-hydroksychinoliny).
2. DEFINICJA
Zawartość chinolin-8-olu i siarczanu bis(8-hydroksychinoliny) w próbce wyrobu oznaczana zgodnie z tą metodą jest wyrażona w procentach masowych chinolin-8-olu.
3. ZASADA
3.1. Identyfikowanie
Identyfikowanie wykonuje się metodą chromatografii cienkowarstwowej.
3.2. Oznaczanie
Oznaczanie przeprowadza się metodą spektrometryczną przez pomiar gęstości optycznej przy 410 nm kompleksu otrzymanego w reakcji z roztworem Fehlinga.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. Chinolin-8-ol.
4.2. Benzen. Ze względu na jego toksyczność należy przedsięwziąć środki ostrożności podczas pracy z benzenem.
4.3. Chloroform.
4.4. Wodny roztwór wodorotlenku sodu, 50% (m/m).
4.5. Pentahydrat siarczanu miedzi.
4.6. Winian sodowo-potasowy.
4.7. M roztwór kwasu solnego.
4.8. 0,5 M roztwór kwasu siarkowego.
4.9. M roztwór wodorotlenku sodu.
4.10. Etanol.
4.11. Butan-1-ol.
4.12. Kwas octowy lodowaty.
4.13. 0,1 M roztwór kwasu solnego.
4.14. Celit 545 lub równoważny.
4.15. Roztwory standardowe
4.15.1. Odważyć 100 mg chinolin-8-olu (4.1) w 100 ml kolbie miarowej. Rozpuścić w małej ilości kwasu siarkowego (4.8). Uzupełnić do kreski roztworem kwasu siarkowego (4.8).
4.15.2. Odważyć 100 mg chinolin-8-olu w 100 ml kolbie miarowej. Rozpuścić w etanolu (4.10). Uzupełnić do kreski etanolem (4.10) i wymieszać.
4.16. Roztwór Fehlinga Roztwór A
Odważyć 7 g pentahydratu siarczanu miedzi (4.5) w 100 ml kolbie miarowej.
Rozpuścić w małej ilości wody. Uzupełnić do kreski wodą i wymieszać.
Roztwór B
Odważyć 35 g winianu sodowo-potasowego (4.6) w 100 ml kolbie miarowej.
Rozpuścić w 50 ml wody. Dodać 20 ml wodorotlenku sodu (4.4). Uzupełnić wodą do kreski i wymieszać. Bezpośrednio przed analizą przenieść po 10 ml roztworu A i roztworu B do 100 ml kolby miarowej. Uzupełnić do kreski i wymieszać.
4.17. Roztwory wymywające do chromatografii cienkowarstwowej
I: butan-1-ol (4.11) / kwas octowy (4.12) / woda (80:20:20; v/v/v)
II: chloroform (4.13) / kwas octowy (4.12) (95:5; v/v).
4.18. 2,6-dichloro-4-(chloroimino)-cykloheksa-2,5-dienon, 1% (m/v) roztwór etanolu (4.10).
4.19. Węglan sodu, 1% (m/v) roztwór wodny.
4.20. Etanol (4.10), 30% (v/v) roztwór wodny.
4.21. Etylenodiaminotetraoctan disodu, 5% (m/v) roztwór wodny.
4.22. Roztwór buforowy o pH = 7
Odważyć 27 g bezwodnego diwodoroortofosforanu potasu i 70 g trihydratu wodoroortofosforanu disodu w jednolitrowej kolbie miarowej. Uzupełnić wodą do kreski.
4.23. Gotowe płytki do chromatografii cienkowarstwowej
Gotowe płytki do chromatografii cienkowarstwowej o grubości 0,25 mm (na przykład Merck Kieselgel 60 lub równoważne). Przed użyciem spryskać 10 ml odczynnika (4.21) i wysuszyć w temperaturze 80°C.
5. APARATURA
5.1. 100 ml kolba okrągłodenna ze szlifem.
5.2. Kolby miarowe.
5.3. Pipety kalibrowane, 10 i 5 ml.
5.4. Pipety ze zbiornikiem gruszkowym, 20, 15, 10 i 5 ml.
5.5. Rozdzielacze, 100, 50 i 25 ml.
5.6. Karbowana bibuła filtracyjna, średnica 90 mm.
5.7. Obrotowy aparat wyparny.
5.8. Chłodnica zwrotna ze szlifem.
5.9. Spektrofotometr.
5.10. Naczynka pomiarowe o długości drogi optycznej 10 mm.
5.11. Mieszadło magnetyczne z ogrzewaniem.
5.12. Wymiary szklanej kolumny chromatograficznej: długość 160 mm, średnica 8 mm, przewężenie w dolnym końcu zawiera tampon z waty szklanej, a w górnym końcu łącznik do podłączenia dopływu zwiększonego ciśnienia.
6. PROCEDURA
6.1. Identyfikowanie
6.1.1. Próbki ciekłe
6.1.1.1. Odczyn części badanej próbki doprowadza się do pH = 7,5 i nanosi się 10 µl na linię bazową przygotowanej uprzednio płytki cienkowarstwowej z żelem krzemionkowym (4.23).
6.1.1.2. 10 i 30 µl roztworu standardowego (4.15.2) nanosi się w dwóch dodatkowych punktach linii początkowej, po czym chromatogram na płytce jest rozwijany w jednym z dwóch roztworów wymywających (4.17).
6.1.1.3. Kiedy czoło rozpuszczalnika osiągnie 150 mm, płytkę suszy się w temperaturze 110°C (w ciągu 15 minut). W świetle lampy UV (366 nm) plamy chinolin-8-olu dają żółtą fluorescencję.
6.1.1.4. Spryskać płytkę roztworem węglanu sodu (4.19). Wysuszyć i spryskać roztworem 2,6-dichloro-4-(chloroimino)-cykloheksa-2,5-dienonu (4.18). Chinolin-8-ol staje się widoczny w postaci niebieskiej plamy.
6.1.2. Próbki stałe lub kremy
6.1.2.1. Zawiesić 1 g próbki w 5 ml roztworu buforowego (4.22). Następnie przenieść z 10 ml chloroformu (4.3) do rozdzielacza i wytrząsnąć. Po oddzieleniu warstwy chloroformowej warstwę wodną ekstrahować jeszcze dwa razy po 10 ml chloroformu (4.3). Odparować połączone i przesączone ekstrakty chloroformowe prawie do sucha w 100 ml kolbie okrągłodennej (5.1) na wyparce obrotowej (5.7). Rozpuścić pozostałość w 2 ml chloroformu (4.3) i nanieść po 10 i 30 µl otrzymanego roztworu na płytkę do chromatografii cienkowarstwowej z żelem krzemionkowym (4.23) i postępować dalej zgodnie z metodą opisaną w pkt 6.1.1.1.
6.1.2.2. Nanieść 10 i 30 µl roztworu standardowego (4.15.2) na płytkę i kontynuować postępowanie zgodnie z opisem w pkt 6.1.1.2–6.1.1.4.
6.2. Oznaczanie
6.2.1. Próbki ciekłe
6.2.1.1. Odważyć 5 g próbki w 100 ml kolbie okrągłodennej. Dodać 1 ml roztworu kwasu siarkowego (4.8) i odparować mieszaninę prawie do sucha pod zmniejszonym ciśnieniem w temperaturze 50°C.
6.2.1.2. Rozpuścić tę pozostałość w 20 ml ciepłej wody. Przenieść do 100 ml kolby miarowej. Przepłukać trzykrotnie 20 ml wody. Uzupełnić do 100 ml wodą i wymieszać.
6.2.1.3. Wprowadzić pipetą 5 ml tego roztworu do 50 ml rozdzielacza (5.5). Dodać 10 ml roztworu Fehlinga (4.16). Ekstrahować otrzymany kompleks chinolin-8-olu miedzi [w ISO: Cu-oksyna] trzykrotnie po 8 ml chloroformu (4.3).
6.2.1.4. Przesączyć i zebrać warstwy chloroformowe w 25 ml kolbie miarowej (5.2). Uzupełnić do kreski chloroformem (4.3) i wytrząsnąć. Zmierzyć gęstość optyczną żółtego roztworu w porównaniu z chloroformem przy 410 nm.
6.2.2. Próbki stałe lub kremy
6.2.2.1. Odważyć 0,500 g próbki w 100 ml kolbie okrągłodennej (4.1). Dodać 30 ml benzenu (4.2) i 20 ml kwasu solnego (4.7). Ogrzewać zawartość kolby do wrzenia pod chłodnicą zwrotną, z mieszaniem w ciągu 30 minut.
6.2.2.2. Przenieść zawartość kolby do 100 ml rozdzielacza (5.5). Popłukać 5 ml 1 N kwasu solnego (4.7). Przenieść fazę wodną do kolby okrągłodennej (5.1) i przemyć warstwę benzenową 5 ml kwasu solnego (4.7).
6.2.2.3. W przypadku emulsji, które utrudniają dalsze działania, zmieszać 0,500 g próbki z 2 g celitu 545 (4.14) do utworzenia sypkiego proszku. Przenieść mieszaninę w małych porcjach na szklaną kolumnę chromatograficzną (5.12).
Po każdym dodaniu mieszaniny ubijać wypełnienie kolumny do dołu. Natychmiast po przeniesieniu całej mieszaniny do kolumny należy wymywać ją kwasem solnym (4.13) w taki sposób, aby otrzymać 10 ml eluatu średnio w ciągu 10 minut (jeżeli to konieczne, elucję można wykonać pod niewielkim ciśnieniem azotu). Podczas eluowania należy się upewnić, że pewna warstwa kwasu solnego zawsze znajduje się powyżej wypełnienia kolumny. Pierwsze 10 ml eluatu jest następnie poddane postępowaniu opisanemu w pkt 6.2.2.4.
6.2.2.4. Odparować zebrane warstwy wodne (6.2.2.2) lub eluaty (6.2.2.3) prawie do sucha w wyparce rotacyjnej pod zmniejszonym ciśnieniem.
6.2.2.5. Rozpuścić pozostałość w 6 ml roztworu wodorotlenku sodu (4.9). Dodać 20 ml roztworu Fehlinga (4.16) i przenieść zawartość kolby do 50 ml rozdzielacza (5.5). Popłukać kolbę 8 ml chloroformu (4.3). Wytrząsnąć i przesączyć warstwę chloroformową do 50 ml kolby miarowej (5.2).
6.2.2.6. Powtórzyć trzykrotnie ekstrakcję chloroformem (4.3). Przesączyć warstwy chloroformowe i zebrać w 50 ml kolbie miarowej. Uzupełnić do kreski chloroformem (4.3) i wytrząsnąć. Zmierzyć gęstość optyczną żółtego roztworu w porównaniu z chloroformem (4.3) przy 410 nm.
7. KRZYWA STANDARDOWA
Do czterech 100 ml kolb okrągłodennych (5.1), każdej zawierającej 3 ml 30% wodnego roztworu etanolu (4.20), dodać pipetą porcje po 5, 10, 15 i 20 ml roztworu standardowego (4.15.1) odpowiadające 5, 10, 15 i 20 mg chinolin-8-olu. Postępować zgodnie z opisem w pkt 6.2.1.
8. OBLICZENIE
8.1. Próbki ciekłe
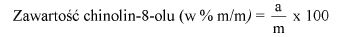
gdzie:
a = ilość miligramów chinolin-8-olu odczytana z krzywej standardowej (7),
m = ilość (w miligramach) próbki analitycznej (6.2.1.1).
8.2. Próbki stałe lub kremy
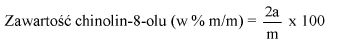
gdzie:
a = ilość miligramów chinolin-8-olu odczytana z krzywej standardowej (7),
m = ilość (w miligramach) próbki analitycznej (6.2.2.1).
9. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości ok. 0,3% chinolin-8-olu różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć wartości absolutnej 0,02%.
XVI. OZNACZANIE AMONIAKU
1. CEL I ZAKRES STOSOWANIA
Celem metody jest oznaczanie wolnego amoniaku w produktach kosmetycznych.
2. DEFINICJA
Zawartość amoniaku w próbce wyrobu jest wyrażona w procentach masowych amoniaku.
3. ZASADA
Roztwór chlorku baru dodaje się do próbki analitycznej produktu kosmetycznego rozcieńczonej w środowisku wodno-metanolowym. Osad, który powstaje, zostaje odsączony lub odwirowany. Ten sposób postępowania pozwala uniknąć strat amoniaku podczas destylacji z parą wodną z pewnych soli amonowych, takich jak węglan lub kwaśny węglan i sole kwasów tłuszczowych, z wyjątkiem octanu amonu. Amoniak jest oddestylowany z przesączu lub z warstwy znad osadu z wirówki z parą wodną i oznaczany miareczkowaniem potencjometrycznym lub innym.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. Metanol.
4.2. Dihydrat chlorku baru, roztwór 25% (m/v).
4.3. Kwas ortoborowy, roztwór 4% (m/v).
4.4. Kwas siarkowy, standardowy 0,25 M roztwór.
4.5. Ciecz przeciwdziałająca pienieniu.
4.6. Wodorotlenek sodu, standardowy 0,5 M roztwór.
4.7. Wskaźnik, jeżeli potrzeba: zmieszać 5 ml 0,1% (m/v) etanolowego roztworu czerwieni metylowej z 2 ml 0,1% (m/v) wodnego roztworu błękitu metylenowego.
5. APARATURA
5.1. Zwykły sprzęt laboratoryjny.
5.2. Wirówka z zamykanymi 100 ml probówkami.
5.3. Aparatura do destylacji z parą wodną.
5.4. Potencjometr.
5.5. Pomiarowa elektroda szklana i elektroda odniesienia z chlorkiem rtęci (kalomelowa).
6. PROCEDURA
6.1. Odważyć do 100 ml kolby miarowej masę (m) próbki odpowiadającej najwyżej 150 mg amoniaku.
6.2. Dodać 10 ml wody, 10 ml metanolu (4.1) i 10 ml roztworu chlorku baru (4.2). Uzupełnić metanolem (4.1) do 100 ml.
6.3. Wymieszać i pozostawić na noc w lodówce (5°C).
6.4. Następnie przesączyć lub odwirować jeszcze zimny roztwór w zamkniętych probówkach na 10 minut tak, aby otrzymać przezroczysty przesącz lub sklarowaną warstwę nad osadem.
6.5. Wprowadzić pipetą 40 ml tego przezroczystego roztworu do aparatury do destylacji z parą wodną (5.3) i następnie dodać, jeżeli potrzeba, 0,5 ml cieczy przeciwdziałającej pienieniu (4.5).
6.6. Destylować i zebrać 200 ml destylatu w 250 ml zlewce zawierającej 10 ml standardowego kwasu siarkowego (4.4) i 0,1 ml wskaźnika (4.7).
6.7. Odmiareczkować nadmiar kwasu standardowym roztworem wodorotlenku sodu (4.6).
6.8. Uwaga: W przypadku oznaczenia potencjometrycznego, zebrać 200 ml destylatu w 250 ml zlewce zawierającej 25 ml roztworu kwasu ortoborowego (4.3) i miareczkować standardowym kwasem siarkowym (4.4), zapisując krzywą neutralizacji.
7. OBLICZENIA
7.1. Obliczanie w przypadku odmiareczkowywania nadmiaru
Przyjęto:
V1 = objętość (w mililitrach) użytego roztworu wodorotlenku sodu (4.6),
M1 = jego rzeczywiste stężenie molowe (4.6),
M2 = oznaczone stężenie molowe roztworu kwasu siarkowego (4.4),
m = masa (w miligramach) pobranej próbki analitycznej (6.1),
zatem:
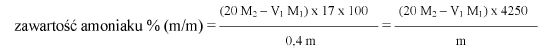
7.2. Obliczanie w przypadku bezpośredniego miareczkowania potencjometrycznego
Przyjęto:
V2 = objętość (w mililitrach) użytego roztworu kwasu siarkowego (4.4),
M2 = jego rzeczywiste stężenie molowe (4.4),
m = masa (w miligramach) pobranej próbki analitycznej (6.1),
zatem:
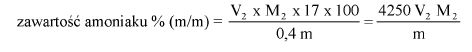
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości amoniaku ok. 6% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,6%.
XVII. IDENTYFIKOWANIE I OZNACZANIE NITROMETANU
1. CEL I ZAKRES STOSOWANIA
Metoda jest odpowiednia do identyfikowania i oznaczania nitrometanu do zawartości ok. 0,3% w produktach kosmetycznych w opakowaniach aerozolowych.
2. DEFINICJA
Zawartość nitrometanu w próbce wyrobu oznaczona zgodnie z tą metodą jest wyrażona w procentach masowych nitrometanu w stosunku do całkowitej zawartości opakowania aerozolowego.
3. ZASADA
Nitrometan jest identyfikowany w reakcji barwnej. Nitrometan jest oznaczany metodą chromatografii gazowej po dodaniu wewnętrznego standardu.
4. IDENTYFIKOWANIE
4.1. Odczynniki
Wszystkie odczynniki powinny być czyste do analizy.
4.1.1. Wodorotlenek sodu, 0,5 M roztwór
4.1.2. Odczynnik Folina
Rozpuścić 0,1 g 3,4-diokso-3,4-dihydronaftaleno-1-sulfonianu sodu w wodzie i rozcieńczyć do 100 ml.
4.2. Procedura
Do 1 ml próbki dodać 10 ml odczynnika wymienionego w pkt 4.1.1 i 1 ml odczynnika wymienionego w pkt 4.1.2. Fioletowe zabarwienie wskazuje na obecność nitrometanu.
5. OZNACZANIE
5.1. Odczynniki
Wszystkie odczynniki powinny być czyste do analizy.
5.1.1. Chloroform (standard wewnętrzny 1).
5.1.2. 2,4-dimetyloheptan (standard wewnętrzny 2).
5.1.3. Etanol, 95%.
5.1.4. Nitrometan.
5.1.5. Chloroformowy roztwór odniesienia
Do wytarowanej 25 ml kolby miarowej wprowadzić 650 mg chloroformu (5.1.1). Dokładnie zważyć ponownie kolbę i zawartość. Uzupełnić do 25 ml 95% etanolem (5.1.3). Zważyć i obliczyć procent masowy chloroformu w tym roztworze.
5.1.6. Roztwór odniesienia 2,4-dimetyloheptanu
Sporządzić w podobny sposób do chloroformowego roztworu odniesienia, lecz zważyć 270 mg 2,4-dimetyloheptanu (5.1.2) w kolbie miarowej o pojemności 25 ml.
5.2. Aparatura
5.2.1. Chromatograf gazowy z detektorem płomieniowo-jonizacyjnym.
5.2.2. Aparatura do pobierania próbek aerozoli (butelka do przenoszenia, łączniki do mikrostrzykawek itp.) jak opisano w części II „Praktyczne przygotowanie próbek laboratoryjnych”.
5.2.3. Zwykły sprzęt laboratoryjny.
5.3. Procedura
5.3.1. Przygotowanie próbki
Do 100 ml wytarowanej butelki do przenoszenia, przepłukanej lub opróżnionej z gazów zgodnie ze sposobem postępowania opisanym w pkt 5.4 w części II „Praktyczne przygotowanie próbek laboratoryjnych”, wprowadzić ok. 5 ml jednego z roztworów standardowych (5.1.5 lub 5.1.6). Stosować szklaną strzykawkę o pojemności 10 lub 20 ml, bez igły, dostosowaną do części łączących według sposobu opisanego w pkt 5 w części II „Praktyczne przygotowanie próbek laboratoryjnych”. Zważyć ponownie dla oznaczenia wprowadzanej ilości. Stosując ten sam sposób, przenieść do tej butelki ok. 50 g zawartości próbki z opakowania aerozolowego. Ponownie zważyć dla określenia ilości przeniesionej próbki. Dobrze wymieszać.
Wprowadzić ok. 10 µl próbki, stosując mikrostrzykawkę (5.2.2). Wykonać pięć wstrzyknięć próbki.
5.3.2. Przygotowanie standardowej próby
W 50 ml kolbie miarowej zważyć dokładnie ok. 500 mg nitrometanu (5.1.4) i 500 mg chloroformu (5.1.1) lub 210 mg 2,4-dimetyloheptanu (5.1.2). Uzupełnić objętość 95% etanolem (5.1.3). Dobrze wymieszać. Umieścić 5 ml tego roztworu w 20 ml kolbie miarowej. Uzupełnić objętość 95% etanolem (5.1.3). Wprowadzić strzykawką ok. 10 µl próbki, stosując mikrostrzykawkę (5.2.2). Wykonać pięć wstrzyknięć próbki.
5.3.3. Warunki chromatografii gazowej. 5.3.3.1. Kolumna
Składa się z dwóch części, pierwszej zawierającej jako wypełnienie ftalan didecylu osadzony na Gas Chrom Q, drugiej zawierającej jako wypełnienie Ucon 50 HB 280X na Gas Chrom Q. Przygotowana połączona kolumna powinna mieć zdolność rozdzielczą „R” równą lub większą niż 1,5, obliczoną według wzoru:
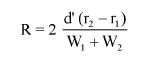
gdzie:
r1 i r2 = czas retencji (w minutach),
W1 i W2 = szerokość pików w połowie wysokości (w milimetrach),
d' = szybkość przesuwu papieru (w milimetrach/minutę).
Jako przykład uzyskano wymaganą rozdzielczość na kolumnie zawierającej następujące dwie części:
Kolumna A:
Materiał: stal nierdzewna
Długość: 1,5 m
Średnica: 3 mm
Wypełnienie: 20% ftalan didecylu na Gas Chrom Q (100–120 mesh)
Kolumna B:
Materiał: stal nierdzewna
Długość: 1,5 m
Średnica: 3 mm
Wypełnienie: 20% Ucon 50 HB 280X na nośniku Gas Chrom Q (100–120 mesh)
5.3.3.2 Detektor
Odpowiedni poziom czułości dla elektrometru detektora płomieniowo-jonizacyjnego wynosi 8 x 10-10A.
5.3.3.3. Warunki temperaturowe
Stwierdzono, że odpowiednie są następujące warunki:
Otwór wlotowy dozownika: 150°C
Detektor: 150°C
Kolumna: między 50 i 80°C zależnie od pojedynczych kolumn i aparatu.
5.3.3.4. Odpowiednie zasilanie gazem
Gaz nośny: azot
Ciśnienie: 2,1 bara
Przepływ: 40 ml/min
Zasilanie detektora: według wskazówek producenta detektora.
6. OBLICZENIA
6.1. Współczynnik reakcji nitrometanu obliczony w stosunku do użytego wewnętrznego standardu.
Jeżeli „n” oznacza nitrometan, przyjęto:
kn = jego współczynnik reakcji,
m'n = jego masa (w gramach) w mieszaninie,
S'n = powierzchnia jego piku.
Jeżeli „c” oznacza standard wewnętrzny, chloroform lub 2,4-dimetyloheptan, przyjęto:
m'c = jego masa (w gramach) w mieszaninie,
S'c = powierzchnia jego piku,
zatem:
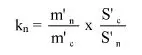
(kn zależy od rodzaju aparatury).
6.2. Stężenie nitrometanu w próbce
Jeżeli „n” oznacza nitrometan, przyjęto:
kn = jego współczynnik reakcji,
Sn = powierzchnia jego piku.
Jeżeli „c” oznacza wewnętrzny standard, chloroform lub 2,4-dimetyloheptan, przyjęto:
mc = jego masa (w gramach) w mieszaninie,
Sc = powierzchnia jego piku,
M = masa (w gramach) przeniesionej próbki aerozolu,
zatem zawartość nitrometanu w próbce wynosi:
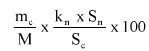
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości nitrometanu ok. 0,3% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć wartości absolutnej 0,03% (m/m).
XVIII. IDENTYFIKOWANIE I OZNACZANIE KWASU MERKAPTOOCTOWEGO W ŚRODKACH DO TRWAŁEJ ONDULACJI, PROSTOWANIA WŁOSÓW I DEPILOWANIA
1. CEL I ZAKRES STOSOWANIA
Celem metody jest identyfikowanie i oznaczanie kwasu merkaptooctowego w środkach do trwałej ondulacji, prostowania włosów i depilowania, w których mogą się znajdować inne środki redukujące.
2. DEFINICJA
Zawartość kwasu merkaptooctowego oznaczona zgodnie z tą metodą jest wyrażona w procentach masowych kwasu merkaptooctowego.
3. ZASADA
Kwas merkaptooctowy identyfikuje się w testach kroplowych i metodą chromatografii cienkowarstwowej i oznacza jodometrycznie lub metodą chromatografii gazowej.
4. IDENTYFIKOWANIE
4.1. Identyfikowanie w testach kroplowych.
4.1.1. Odczynniki
Wszystkie odczynniki powinny być czyste do analizy.
4.1.1.1. Bibuła z dioctanem ołowiu.
4.1.1.2. Roztwór kwasu solnego (jedna objętość stężonego kwasu solnego i jedna objętość wody).
4.1.2. Procedura
4.1.2.1. Identyfikowanie kwasu merkaptooctowego przez barwną reakcję z dioctanem ołowiu (II).
Umieścić kroplę analizowanej próbki na bibule z dioctanem ołowiu (II) (4.1.1.1). Jeżeli pojawi się intensywny żółty kolor, oznacza to, że kwas merkaptooctowy jest prawdopodobnie obecny. Czułość: 0,5%.
4.1.2.2. Wykrywanie siarczków nieorganicznych w reakcji wydzielania siarkowodoru podczas zakwaszania próbki.
Wprowadzić do probówki kilka miligramów badanej próbki. Dodać 2 ml wody destylowanej i 1 ml kwasu solnego (4.1.1.2). Wydziela się siarkowodór rozpoznawalny po zapachu i czarny osad siarczku ołowiu powstaje na bibule z dioctanem ołowiu (II) (4.1.1.1). Czułość: 50 ppm.
4.1.2.3. Wykrywanie siarczynów w reakcji powstawania ditlenku siarki po zakwaszeniu próbki.
Postępować według opisu w pkt 4.1.2.2. Doprowadzić do wrzenia. Ditlenek siarki jest rozpoznawalny po zapachu i dzięki własnościom redukującym, na przykład wobec jonów nadmanganianowych.
4.2. Identyfikowanie przez chromatografię cienkowarstwową.
4.2.1. Odczynniki
Wszystkie odczynniki powinny być czyste do analizy.
4.2.1.1. Kwas merkaptooctowy (kwas tioglikolowy) o czystości co najmniej 98% oznaczonej jodometrycznie.
4.2.1.2. Kwas 2,2'-ditiodi(octowy) o czystości co najmniej 99% oznaczonej jodometrycznie.
4.2.1.3. Kwas 2-merkaptopropionowy (kwas tiomlekowy) o czystości co najmniej 95% oznaczonej jodometrycznie.
4.2.1.4. Kwas 3-merkaptopropionowy o czystości co najmniej 98% oznaczonej jodometrycznie.
4.2.1.5. 3-merkaptopropano-1,2-diol (1-tioglicerol) o czystości co najmniej 98% oznaczonej jodometrycznie.
4.2.1.6. Płytki cienkowarstwowe, z żelem krzemionkowym, przygotowane fabrycznie, o grubości 0,25 mm.
4.2.1.7. Płytki cienkowarstwowe, z tlenkiem glinu, Merck F 254 E lub równoważne.
4.2.1.8. Stężony kwas solny, d
4.2.1.9. Octan etylu.
4.2.1.10. Chloroform.
4.2.1.11. Eter diizopropylowy.
4.2.1.12. Czterochlorek węgla.
4.2.1.13. Kwas octowy lodowaty.
4.2.1.14. Jodek potasu, 1% (m/v) roztwór wodny.
4.2.1.15. Chlorek platyny (IV), 0,1% (m/v), roztwór wodny.
4.2.1.16. Rozpuszczalniki wymywające
4.2.1.16.1. Octan etylu (4.2.1.9), chloroform (4.2.1.10), eter diizopropylowy (4.2.1.11), kwas octowy (4.2.1.13) (20:20:10:10 objętościowo).
4.2.1.16.2. Chloroform (4.2.1.10), kwas octowy (4.2.1.13) (90:20 objętościowo).
4.2.1.17. Odczynniki wywołujące
4.2.1.17.1. Wymieszać bezpośrednio przed użyciem jednakowe objętości roztworu (4.2.1.14) i roztworu (4.2.1.15).
4.2.1.17.2. Roztwór bromu 5% (m/v)
Rozpuścić 5 g bromu w 100 ml czterochlorku węgla (4.2.1.12).
4.2.1.17.3. Roztwór fluoresceiny 0,1% (m/v)
Rozpuścić 100 mg fluoresceiny w 100 ml etanolu.
4.2.1.17.4. Heptamolibdenian (VI) heksaamonu, 10% (m/v) roztwór wodny.
4.2.1.18. Roztwory odniesienia
4.2.1.18.1. Kwas merkaptooctowy (4.2.1.1), 0,4% (m/v) roztwór wodny.
4.2.1.18.2. Kwas 2,2'-ditiodi(octowy) (4.2.1.2), 0,4% (m/v) roztwór wodny.
4.2.1.18.3. Kwas 2-merkaptopropionowy (4.2.1.3), 0,4% (m/v) roztwór wodny.
4.2.1.18.4. Kwas 3-merkaptopropionowy (4.2.1.4), 0,4% (m/v) roztwór wodny.
4.2.1.18.5. 3-merkaptopropano-1,2-diol (4.2.1.5), 0,4% (m/v) roztwór wodny.
4.2.2. Aparatura
Zwykła aparatura do chromatografii cienkowarstwowej.
4.2.3. Procedura
4.2.3.1. Postępowanie z próbkami
Zakwasić do pH = 1 kilkoma kroplami kwasu solnego (4.2.1.8) i przesączyć, jeżeli to konieczne.
W niektórych przypadkach rozcieńczenie próbki może być pożądane. Jeżeli tak, to przed rozcieńczeniem zakwasić ją kwasem solnym.
4.2.3.2. Rozwijanie chromatogramu (eluowanie)
Umieścić na płytce 1 µl roztworu próbki (4.2.3.1) i po 1 µl każdego z pięciu roztworów odniesienia (4.2.1.18). Wysuszyć starannie w łagodnym strumieniu azotu i eluować płytki rozpuszczalnikami (4.2.1.16.1 lub 4.2.1.16.2). Wysuszyć płytkę możliwie najszybciej, aby uniknąć utleniania tioli.
4.2.3.3. Wykrywanie
Spryskać płytkę jednym z trzech reagentów (4.2.1.17.1, 4.2.1.17.3 lub 4.2.1.17.4). Jeżeli płytkę spryskano odczynnikiem (4.2.1.17.3), należy następnie poddać ją działaniu pary bromu (na przykład w komorze zawierającej małą zlewkę odczynnika (4.2.1.17.2), do chwili, w której plamy staną się widoczne. Wykrywanie przez spryskanie odczynnikiem (4.2.1.17.4) będzie zadawalające tylko wtedy, jeżeli czas suszenia cienkiej warstwy nie przekroczy 30 minut.
4.2.3.4. Interpretacja
Porównać wartości Rf i kolory roztworów odniesienia z odpowiednimi wartościami dla roztworów standardowych. Średnie wartości Rf podane poniżej jako ogólna wskazówka mają tylko wartość porównawczą. Zależą one od:
1) stanu aktywowania cienkiej warstwy w czasie analizy chromatograficznej;
2) temperatury komory chromatograficznej.
Przykładowe wartości Rf otrzymane na płytce z warstwą żelu krzemionkowego
|
| Roztwory rozwijające | |
|
| 4.2.1.16.1 | 4.2.1.16.2 |
| Kwas merkaptooctowy | 0,25 | 0,80 |
| Kwas 2-merkaptopropionowy | 0,40 | 0,95 |
| Kwas 2,2'-ditiodi(octowy) | 0,00 | 0,35 |
| Kwas 3-merkaptopropionowy | 0,45 | 0,95 |
| 3-merkaptopropano-1,2-diol | 0,45 | 0,35 |
5. OZNACZANIE
Oznaczenie kwasu merkaptooctowego musi być wykonane z nieużywanego wyrobu ze świeżo otwartego opakowania w celu zapobieżenia utlenianiu. Oznaczanie powinno się zawsze rozpocząć od metody jodometrycznej (jodometrii).
5.1. Jodometria
5.1.1. Zasada
Oznaczanie jest wykonywane przez utlenianie grupy „-SH” jodem w środowisku kwaśnym, zgodnie z równaniem:
2 HOOC-CH2SH + I2 ® (HOOC-CH2-S)2 + 2I + 2H+
5.1.2. Odczynniki
Jod, 0,05 M roztwór standardowy.
5.1.3. Aparatura
Zwykły sprzęt laboratoryjny.
5.1.4. Procedura
Odważyć dokładnie ilość próbki między 0,5 i 1 g w 150 ml kolbie stożkowej zamykanej korkiem zawierającej 50 ml wody destylowanej. Dodać 5 ml kwasu solnego (4.1.1.2) (pH roztworu ok. 0) i miareczkować roztworem jodu (5.1.2) aż do pojawienia się żółtego koloru. Jeżeli trzeba użyć wskaźnika, to na przykład roztworu skrobi lub czterochlorku węgla.
5.1.5. Obliczenie
Zawartość kwasu merkaptooctowego oblicza się zgodnie z wzorem:
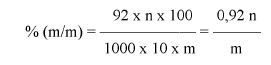
gdzie:
m = masa (w gramach) próbki analitycznej,
n = objętość użytego roztworu jodu (5.1.2).
5.1.6. Uwagi
Jeżeli wynik obliczony jako zawartość kwasu merkaptooctowego wynosi 0,1% lub znacznie poniżej dozwolonego maksymalnego stężenia, nie ma żadnego powodu dla przeprowadzania następnego oznaczania. Jeżeli wynik jest równy maksymalnemu stężeniu lub wyższy od dozwolonego maksymalnego stężenia i identyfikowanie ujawniło obecność kilku środków redukujących, konieczne jest wykonanie oznaczenia metodą chromatografii gazowej.
5.2. Chromatografia gazowa
5.2.1. Zasada
Kwas merkaptooctowy zostaje wydzielony ze środowiska próbki przez wytrącanie roztworem dioctanu kadmu. Po metylowaniu diazometanem, przygotowanym in situ lub lepiej w roztworze eteru dietylowego, pochodna metylowa kwasu merkaptooctowego jest oznaczana chromatografią gazową/cieczową z użyciem kaprylanu metylu jako wewnętrznego standardu.
5.2.2. Odczynniki
Wszystkie odczynniki muszą być czyste do analizy.
5.2.2.1. Kwas merkaptooctowy, 98%.
5.2.2.2. Kwas solny, d
5.2.2.3. Metanol.
5.2.2.4. Dihydrat dioctanu kadmu (II), 10% (m/v) roztwór wodny.
5.2.2.5. Kaprylan metylu, 2% (m/v) roztwór w metanolu.
5.2.2.6. Octanowy roztwór buforowy (pH = 5).
Trihydrat octanu sodu, 77 g.
Kwas octowy lodowaty, 27,5 g.
Woda demineralizowana do otrzymania końcowej objętości jednego litra.
5.2.2.7. Kwas solny, 3 M roztwór w metanolu (5.2.2.3), świeżo przygotowany.
5.2.2.8. 1-metylo-3-nitro-1-nitrozoguanidyna.
5.2.2.9. Wodorotlenek sodu, roztwór 5 M.
5.2.2.10. Jod, 0,05 M roztwór standardowy.
5.2.2.11. Eter dietylowy.
5.2.2.12. Roztwór diazometanu przygotowany z N-metylo-N-nitrozo-4-tolulenosulfonoamidu Otrzymany roztwór zawiera ok. 1,5 g diazometanu w 100 ml eteru dietylowego. Ponieważ diazometan jest gazem toksycznym i bardzo nietrwałym, wszystkie doświadczenia muszą być prowadzone pod sprawnie działającym wyciągiem i należy unikać stosowania aparatury szklanej z połączeniami szlifowymi (istnieją specjalne zestawy przeznaczone do posługiwania się diazometanem).
5.2.3. Aparatura
5.2.3.1. Zwykły sprzęt laboratoryjny.
5.2.3.2. Aparatura do przygotowania diazometanu do metylowania in situ.
5.2.3.3. Aparatura do ulepszonego przygotowania diazometanu (Fieser).
5.2.4. Przygotowanie próbki
Zważyć dokładnie w 50 ml probówce wirówki wystarczającą ilość próbki, aby otrzymać przypuszczalną ilość 50–70 mg kwasu merkaptooctowego. Dodać kilka kropel kwasu solnego (5.2.2.2) do otrzymania pH ok. 3.
Dodać 5 ml demineralizowanej wody i 10 ml roztworu buforu octanowego (5.2.2.6).
Sprawdzić papierkiem wskaźnikowym, czy pH wynosi ok. 5. Następnie dodać 5 ml roztworu dioctanu kadmu (5.2.2.4).
Poczekać 10 minut i następnie odwirować w ciągu co najmniej 15 minut przy 4000 obrotów/min. Usunąć pływającą na wierzchu ciecz, która może zawierać nierozpuszczalny tłuszcz (w przypadku kremów). Tłuszcz ten nie powinien być zmieszany z tiolami, które zbierają się w zgęszczonej masie na dnie probówki.
Sprawdzić, czy nie wytrąca się żaden osad po dodaniu kilku kropel roztworu octanu kadmu (II) (5.2.2.4) do warstwy powierzchniowej.
Jeżeli wcześniej podczas identyfikowania nie stwierdzono żadnych czynników redukujących innych niż tiole, należy sprawdzić metodą jodometryczną, czy zawartość tiolu obecnego w warstwie powierzchniowej nie przekracza 6–8% jego początkowej ilości.
Wprowadzić 10 ml metanolu (5.2.2.3) do probówki wirówki zawierającej osad i starannie rozproszyć osad, mieszając. Odwirować ponownie w ciągu co najmniej 15 minut przy 4000 obrotów/min. Zlać ciecz pływającą na wierzchu i sprawdzić, czy są obecne lub nieobecne tiole. Przemywać osad po raz drugi, postępując w ten sam sposób. Używając stale tej samej probówki wirówki, dodać:
1) 2 ml roztworu kaprylanu metylu (5.2.2.5);
2) 5 ml kwasu solnego w metanolu (5.2.2.7).
Całkowicie rozpuścić tiole (niewielka ilość nierozpuszczonej substancji może pozostać z domieszek). Jest to roztwór „S”.
Pobrać część tego roztworu, aby potwierdzić jodometrycznie, że zawartość tioli stanowi co najmniej 90% zawartości otrzymanej zgodnie z pkt 5.1.
5.2.5. Metylowanie
Metylowanie jest prowadzone metodą in situ (5.2.5.1) lub za pomocą wcześniej przygotowanego roztworu diazometanu (5.2.5.2).
5.2.5.1. Metylowanie in situ
Do aparatu do metylowania (5.2.3.2) zawierającego 1 ml eteru (5.2.2.11) wprowadzić 50 µl roztworu „S” i metylować metodą (5.2.3.2) z dodatkiem ok. 300 mg 1-metylo-3-nitro-1-nitrozoguanidyny (5.2.2.8). Po 15 minutach (roztwór eterowy powinien być żółty, co wskazuje na nadmiar diazometanu) przenieść próbkę roztworu do 2 ml butelki z hermetycznym korkiem. Umieścić w lodówce do następnego dnia. Metylować równocześnie dwie próbki.
5.2.5.2. Metylowanie przygotowanym wcześniej roztworem diazometanu.
Do 5 ml kolby z korkiem wprowadzić 1 ml roztworu diazometanu (5.2.2.12), następnie 50 µl roztworu „S”. Pozostawić w lodówce do następnego dnia.
5.2.6. Przygotowanie roztworu standardowego
Przygotować standardowy roztwór kwasu merkaptooctowego (5.2.2.1) o znanym stężeniu zawierający ok. 60 mg czystego kwasu merkaptooctowego (5.2.2.1) w 2 ml. Jest to roztwór „E”. Wytrącić osad, zanalizować i metylować tak, jak opisano w pkt 5.2.4 i 5.2.5.
5.2.7. Warunki chromatografii gazowej
5.2.7.1. Kolumna:
Typ: stal nierdzewna
Długość 2 m
Średnica 3 mm
5.2.7.2. Wypełnienie:
20% ftalan didecylu / Chromosorb WAW 80–100 mesh
5.2.7.3. Detektor
Detektor płomieniowo-jonizacyjny. Odpowiednia czułość ustalona dla elektrometru detektora płomieniowo-jonizacyjnego wynosi 8 x 10-10A.
5.2.7.4. Zasilanie gazami:
Gaz nośny: azot o ciśnieniu 2,2 bara i przepływie 35 ml/min
Gaz pomocniczy: wodór o ciśnieniu 1,8 bara i przepływie 15 ml/min
Zasilanie detektora takie, jak podane przez producenta aparatu.
5.2.7.5. Warunki temperaturowe
Dozownik: 200°C
Detektor: 200ºC
Kolumna: 90°C
5.2.7.6. Szybkość przesuwu papieru rejestratora: 5 mm/min.
5.2.7.7. Wprowadzana ilość próbki: 3 µl; wykonać 5 powtórzeń wprowadzania próbki.
5.2.7.8. Warunki chromatografii gazowej są podane orientacyjnie. Pozwalają one na uzyskanie rozdzielczości „R” równej lub wyższej od 1,5, zgodnie z wzorem:
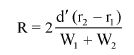
gdzie:
r1 i r2 = czas retencji (w minutach),
W1 i W2 = szerokość pików w połowie wysokości (w milimetrach),
d' = szybkość przesuwu papieru (w milimetrach na minutę).
Poleca się stosowanie programu wzrostu temperatury przez jej regulację w zakresie od 90 do 150°C z szybkością 10°C na minutę tak, aby wyeliminować substancje, które mogą przeszkadzać w kolejnych pomiarach.
5.2.8. Obliczenia
5.2.8.1. Współczynnik korekcyjny kwasu merkaptooctowego.
Jest on obliczany w stosunku do kaprylanu metylu na podstawie mieszaniny standardowej.
Jeżeli „t” oznacza kwas merkaptooctowy, przyjęto:
kt = jego współczynnik reakcji,
m't = jego masę (w miligramach) w mieszaninie,
S't = powierzchnię jego piku.
Jeżeli „c” oznacza kaprylan metylu, przyjęto:
m'c = jego masę (w miligramach) w mieszaninie,
S'c = powierzchnię jego piku,
zatem:
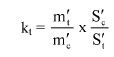
Współczynnik zmienia się zależnie od stosowanej aparatury.
5.2.8.2. Zawartość kwasu merkaptooctowego obecnego w próbce
Jeżeli „t” oznacza kwas merkaptooctowy, przyjęto:
kt = jego współczynnik reakcji,
St = jego powierzchnię piku.
Jeżeli „c” oznacza kaprylan metylu, przyjęto:
mc = masę kaprylanu metylu (w miligramach) w mieszaninie,
Sc = powierzchnię jego piku,
M = masę (w miligramach) początkowej próbki analitycznej,
zatem zawartość kwasu merkaptooctowego obecnego w próbce wynosi:
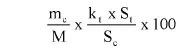
6. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości kwasu merkaptooctowego 8% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,8% (m/m).
XIX. IDENTYFIKOWANIE I OZNACZANIE HEKSACHLOROFENU
A. IDENTYFIKOWANIE
1. CEL I ZAKRES STOSOWANIA
Metoda właściwa do badań wszystkich produktów kosmetycznych.
2. ZASADA
Heksachlorofen w próbce wyrobu jest ekstrahowany octanem etylu i identyfikowany metodą chromatografii cienkowarstwowej.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. Kwas siarkowy, roztwór 4 M.
3.2. Celit AW.
3.3. Octan etylu.
3.4. Rozpuszczalnik rozwijający: benzen zawierający 1% (v/v) kwasu octowego lodowatego.
3.5. Środek wywołujący I
Roztwór rodaminy B: rozpuścić 100 mg rodaminy B w mieszaninie 150 eteru dietylowego, 70 ml absolutnego etanolu i 16 ml wody.
3.6. Środek wywołujący II
Roztwór 2,6-dibromo-4-(chloroimino)cykloheksa-2,5-dienonu: rozpuścić 400 mg 2,6-dibromo-4-(chloroimino)cykloheksa-2,5-dienonu w 100 ml metanolu (przygotować codziennie świeży roztwór).
Roztwór węglanu sodu: rozpuścić 10 g węglanu sodu w 100 ml demineralizowanej wody.
3.7. Roztwór odniesienia
Heksachlorofen, 0,05% (m/v), roztwór w octanie etylu.
4. APARATURA
4.1. Płytki z żelem krzemionkowym Kiesel gel 254 TLC, 200 x 200 mm (lub równoważne).
4.2. Zwykły sprzęt do chromatografii cienkowarstwowej.
4.3. Łaźnia termostatowana w temperaturze 26°C do utrzymywania komory chromatograficznej w stałej temperaturze.
5. PRZYGOTOWANIE PRÓBKI DO BADAŃ
5.1. Starannie wymieszać 1 g homogenizowanej próbki z 1 g Celit AW (3.2) i 1 ml kwasu siarkowego (3.1).
5.2. Suszyć w temperaturze 100°C przez dwie godziny.
5.3. Schłodzić i dokładnie sproszkować wysuszoną pozostałość.
5.4. Ekstrahować dwukrotnie 10 ml octanu etylu (3.3), odwirowując po każdej ekstrakcji, i połączyć warstwy octanu etylu.
5.5. Odparować w temperaturze 60°C.
5.6. Rozpuścić pozostałość w 2 ml octanu etylu (3.3).
6. PROCEDURA
6.1. Nanieść 2 µl roztworu próbki analitycznej (5.6) i 2 µl roztworu odniesienia (3.7) na płytkę do chromatografii cienkowarstwowej (4.1).
6.2. Nasycić komorę (4.3) rozpuszczalnikiem rozwijającym (3.4).
6.3. Umieścić płytkę chromatograficzną w komorze i rozwijać do wysokości 150 mm.
6.4. Wyjąć płytkę chromatograficzną i wysuszyć w wentylowanej suszarce w temperaturze ok. 105°C.
6.5. Wywoływanie
Plamy heksachlorofenu na płytce cienkowarstwowej wywołuje się według opisu w pkt 6.5.1 lub 6.5.2.
6.5.1. Spryskać płytkę równomiernie roztworem wywołującym I (3.5). Po 30 minutach oceniać płytkę w świetle UV przy 254 nm.
6.5.2. Spryskać płytkę środkiem wywołującym II roztworem 2,6-dibromo-4-(chloroimino)cykloheksa-2,5-dienonu (3.6). Następnie spryskać płytkę roztworem węglanu sodu (3.6). Oceniać płytkę w świetle dziennym po 10 minutach suszenia w temperaturze pokojowej.
7. INTERPRETACJA
7.1. Środek wywołujący I (3.5)
Heksachlorofen pojawia się jako niebieskawa plama na żółto-pomarańczowym tle i ma współczynnik Rf w przybliżeniu 0,5.
7.2. Środek wywołujący II (3.6)
Heksachlorofen pojawia się jako plama szafirowo-turkusowa na białym tle i ma współczynnik Rf w przybliżeniu 0,5.
B. OZNACZANIE
1. CEL I ZAKRES STOSOWANIA
Metoda może być stosowana do badania wszystkich produktów kosmetycznych.
2. DEFINICJA
Zawartość heksachlorofenu w próbce wyrobu oznaczona zgodnie z tą metodą jest wyrażona w procentach masowych heksachlorofenu.
3. ZASADA
Heksachlorofen jest oznaczany po przeprowadzeniu w pochodną metylową metodą chromatografii gazowej z detektorem wychwytu elektronów.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. Octan etylu.
4.2. N-metylo-N-nitrozo-4-toluenosulfonoamid (diazald).
4.3. Eter dietylowy.
4.4. Metanol.
4.5. 2-(2-etoksyetoksy)etanol (karbitol).
4.6. Kwas mrówkowy.
4.7. Wodorotlenek potasu, 50% (m/m) roztwór wodny (przygotowany świeżo każdego dnia).
4.8. Heksan do spektroskopii.
4.9. Bromochlorofen (substancja standardowa nr 1).
4.10. 4,4',6,6'-tetrachloro-2,2'-tiodifenol (substancja standardowa nr 2).
4.11. Eter 2,4,4'-trichloro-2-hydroksydifenylowy (substancja standardowa nr 3).
4.12. Aceton.
4.13. 4 M kwas siarkowy.
4.14. Celit AW.
4.15. Roztwór kwasu mrówkowego / octanu etylu, 10% (v/v).
4.16. Heksachlorofen.
5. APARATURA
5.1. Zwykły sprzęt laboratoryjny.
5.2. Miniaparat do otrzymywania diazometanu.
5.3. Chromatograf gazowy wyposażony w detektor wychwytowy elektronów ze źródłem 63 Ni.
6. PROCEDURA
6.1. Przygotowanie roztworu standardowego
Wybiera się taką substancję standardową, która nie koliduje z żadnym składnikiem zawartym w składzie analizowanego produktu. Zwykle substancja standardowa nr 1 jest najodpowiedniejsza (4.9).
6.1.1. Odważyć dokładnie ok. 50 mg substancji standardowej nr 1, 2 lub 3 (4.9, 4.10 lub 4.11) i 50 mg heksachlorofenu (4.16) do 100 ml kolby miarowej. Uzupełnić objętość do kreski octanem etylu (4.1) (roztwór A). Rozcieńczyć 10 ml roztworu A do 100 ml octanem etylu (4.1) (roztwór B).
6.1.2. Odważyć dokładnie ok. 50 mg substancji standardowej nr 1, 2 lub 3 (4.9, 4.10 lub 4.11) do 100 ml kolby miarowej. Uzupełnić objętość do kreski octanem etylu (4.1) (roztwór C).
6.2. Przygotowanie próbki1)
Odważyć dokładnie 1 g ujednoliconej próbki i wymieszać starannie z 1 ml kwasu siarkowego (4.13), 15 ml acetonu (4.12) i 8 g Celitu AW (4.14). Suszyć mieszaninę na powietrzu w ciągu 30 minut w łaźni parowej, następnie suszyć w ciągu półtorej godziny w suszarce z przepływem powietrza. Schłodzić, rozetrzeć pozostałość na proszek i nanieść na szklaną kolumnę. Eluować octanem etylu (4.1) i zebrać 100 ml. Dodać 2 ml roztworu wewnętrznego standardu (roztwór C) (6.1.2).
6.3. Metylowanie próbki
Schłodzić wszystkie odczynniki oraz aparaturę do temperatury między 0 i 4ºC na dwie godziny. W zewnętrznej komorze aparatury do diazometanu umieścić 1,2 ml roztworu otrzymanego według opisu w pkt 6.2 i 0,1 ml metanolu (4.4). W centralnym zbiorniku umieścić ok. 200 mg diazaldu (4.2), dodać 1 ml karbitolu (4.5) i 1 ml eteru dietylowego (4.3) i rozpuścić. Złożyć aparaturę, w połowie zanurzyć aparaturę w łaźni o temperaturze 0°C i wprowadzić strzykawką ok. 1 ml schłodzonego roztworu wodorotlenku sodowego (4.7) do centralnego zbiornika. Upewnić się, że żółty kolor powstały przy tworzeniu się diazometanu jest trwały. Jeżeli żółty kolor nie jest trwały, należy powtórzyć metylowanie, dodając następną porcję 200 mg diazaldu (4.2).2)
Aparaturę usuwa się z łaźni po 15 minutach i następnie pozostawia zamkniętą w temperaturze otoczenia przez 12 godzin. Otworzyć aparaturę, związać nadmiar diazometanu, dodając kilka kropli 10% (v/v) roztworu kwasu mrówkowego w octanie etylu (4.15) i przenieść roztwór organiczny do 25 ml kolby miarowej. Uzupełnić objętość heksanem (4.8) do kreski.
Wprowadzić strzykawką 1,5 µl tego roztworu do chromatografu.
6.4. Metylowanie próbki standardowej
Schłodzić wszystkie odczynniki oraz aparaturę do temperatury między 0 i 4°C na dwie godziny. W zewnętrznej komorze aparatury do diazometanu umieścić:
0,2 ml roztworu B (6.1.1),
1 ml octanu etylu (4.1),
0,1 ml metanolu (4.4).
Prowadzić metylowanie tak, jak opisano w pkt 6.3. Wprowadzić strzykawką 1,5 µl otrzymanego roztworu do chromatografu.
7. CHROMATOGRAFIA GAZOWA
Kolumna musi zapewnić zdolność rozdzielczą „R” równą 1,5 lub większą, zgodnie z wzorem:
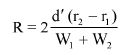
gdzie:
r1 i r2 = czas retencji (w minutach),
W1 i W2 = szerokość pików w połowie wysokości (w milimetrach),
d' = szybkość przesuwu papieru (w milimetrach na minutę).
Następujące warunki chromatografii gazowej uznano za odpowiednie:
Kolumna: stal nierdzewna
Długość: 1,7 m
Średnica: 3 mm
Nośnik:
Chromosorb: WAW
analiza sitowa: 80–120 oczek
Faza stacjonarna: 10% OV 17
Temperatura:
kolumna: 280°C
dozownik: 280°C
detektor: 280°C
Gaz nośny: azot wolny od tlenu
Ciśnienie: 2,3 bara
Przepływ: 30 ml/min
8. OBLICZENIE
8.1. Współczynnik reakcji heksachlorofenu
Jest on obliczany w odniesieniu do wybranej substancji standardowej w stosunku do standardowej mieszaniny.
Przyjęto:
h = heksachlorofen,
kh = jego współczynnik reakcji,
m'h = jego masa (w gramach) w mieszaninie,
A'h = powierzchnia jego piku,
s = wybrana substancja standardowa,
m's = jej masa (w gramach) w mieszaninie,
A's = powierzchnia jej piku.
Współczynnik reakcji heksachlorofenu oblicza się według wzoru:
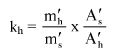
8.2. Ilość heksachlorofenu w próbce Przyjęto:
h = heksachlorofen,
kh = jego współczynnik reakcji,
Ah = powierzchnia jego piku,
s = wybrana substancja standardowa,
ms = jego masa (w gramach) w mieszaninie,
As = powierzchnia jego piku,
M = masa (w gramach) pobranej próbki,
zatem % (m/m) heksachlorofenu w próbce oblicza się według wzoru:
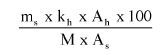
9. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości heksachlorofenu 0,1% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,005% (m/m).
XX. ILOŚCIOWE OZNACZANIE SOLI SODOWEJ N-CHLORO-4-TOLUENOSULFONOAMIDU (INN) (CHLORAMINY-T)
1. CEL I ZAKRES STOSOWANIA
Celem metody jest ilościowe oznaczanie chromatografią cienkowarstwową soli sodowej N-chloro-4-toluenosulfonoamidu (chloramina T) w produktach kosmetycznych.
2. DEFINICJA
Zawartość chloraminy-T w próbce wyrobu oznaczona zgodnie z tą metodą jest wyrażona w procentach masowych (m/m).
3. ZASADA
Chloramina-T ulega całkowitej hydrolizie do 4-toluenosulfonoamidu przez ogrzewanie do wrzenia z kwasem solnym.
Ilość powstałego 4-toluenosulfonoamidu jest oznaczana fotodensytometrycznie przez chromatografię cienkowarstwową.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. Sól sodowa N-chloro-4-toluenosulfonoamid (chloramina-T).
4.2. Standardowy roztwór 4-toluenosulfonoamidu: 50 mg 4-toluenosulfonoamidu w 100 ml etanolu (4.5).
4.3. Kwas solny, 37% (m/m), d
4.4. Eter dietylowy.
4.5. Etanol, 96% (v/v).
4.6. Roztwór rozwijający.
4.6.1. 1-Butanol/etanol (4.5)/woda (40:4:9, v/v/v) lub
4.6.2. Chloroform/aceton (6:4, v/v).
4.7. Przygotowane fabrycznie płytki do chromatografii cienkowarstwowej, żel krzemionkowy 60, bez wskaźnika fluorescencyjnego.
4.8. Nadmanganian potasu.
4.9. Kwas solny, 15% (m/m).
4.10. Odczynnik wywołujący: 2-toluidyna, 1% (m/v) roztwór etanolu (4.5).
5. APARATURA
5.1. Zwykła aparatura laboratoryjna.
5.2. Zwykły sprzęt do chromatografii cienkowarstwowej.
5.3. Fotodensytometr.
6. PROCEDURA
6.1. Hydroliza
Odważyć dokładnie w 50 ml okrągłodennej kolbie ok. 1 g próbki (m). Dodać 5 ml wody i 5 ml kwasu solnego (4.3) i utrzymywać w stanie wrzenia przez jedną godzinę, stosując chłodnicę zwrotną. Bezzwłocznie przenieść gorącą suspensję z wodą do 50 ml kolby miarowej. Pozostawić do schłodzenia i uzupełnić do kreski wodą. Odwirowywać przy co najmniej 3000 obrotów/min przez pięć minut i przefiltrować ciecz pływającą na wierzchu.
6.2. Ekstrakcja
6.2.1. Pobrać 30 ml przesączu i ekstrahować trzykrotnie po 15 ml eteru dietylowego (4.4). Jeżeli to konieczne, wysuszyć warstwy eterowe i połączyć je w 50 ml kolbie miarowej. Uzupełnić eterem dietylowym (4.4).
6.2.2. Pobrać 25 ml wysuszonego ekstraktu eterowego i odparować do sucha w strumieniu azotu. Ponownie rozpuścić pozostałość w 1 ml etanolu (4.5).
6.3. Chromatografia cienkowarstwowa
6.3.1. Nanieść 20 µl pozostałości etanolowej (6.2) na płytkę do chromatografii cienkowarstwowej (4.7).
W tym samym czasie i w ten sam sposób nanieść po 8, 12, 16 i 20 µl standardowego roztworu 4-toluenosulfonoamidu (4.2).
6.3.2. Następnie rozwijać w przybliżeniu na wysokość 150 mm w roztworze rozwijającym (4.6.1 lub 4.6.2).
6.3.3. Po całkowitym odparowaniu rozpuszczalnika rozwijającego umieścić płytkę na dwie lub trzy minuty w atmosferze pary chloru, utworzonej przez nalanie ok. 100 ml kwasu solnego (4.9) na ok. 2 g nadmanganianu potasu (4.8) w zamkniętym naczyniu. Usunąć nadmiar chloru przez ogrzewanie płytki do temperatury 100°C w ciągu pięciu minut. Następnie spryskać płytkę odczynnikiem wywołującym (4.10).
6.4. Pomiar
Po upływie około jednej godziny zmierzyć fioletowe plamy za pomocą fotodensytometru przy 525 nm.
6.5. Wykreślanie krzywych kalibracji
Wykreślić maksymalne wartości wysokości pików ustalone dla czterech plam 4-toluenosulfonoamidu w porównaniu z odpowiednimi ilościami 4-toluenosulfonoamidu (to jest 4, 6, 8, 10 µg 4-toluenosulfonoamidu na plamę).
7. UWAGA
Metodę można sprawdzić, stosując roztwór 0,1 do 0,2% (m/v) chloraminy-T poddany takiemu samemu postępowaniu jak próbka (6).
8. OBLICZENIE
Zawartość chloraminy-T w próbce wyrażona w procentach masowych jest obliczona następująco:

gdzie:
1,33 = współczynnik konwersji (przemiany) 4-toluenosulfonoamidu (chloraminy-T),
a = ilość (w µg) 4-toluenosulfonoamidu w próbce odczytana z krzywej kalibracyjnej,
m = masa (w gramach) pobranej próbki.
9. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości chloraminy-T ok. 0,2% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,03% (m/m).
XXI. OZNACZANIE CAŁKOWITEGO FLUORU W PASTACH DO ZĘBÓW
1. CEL I ZAKRES STOSOWANIA
Metoda jest przeznaczona do oznaczania całkowitej zawartości fluoru w pastach do zębów. Jest ona odpowiednia dla stężeń nieprzekraczających 0,25%.
2. DEFINICJA
Zawartość fluoru w próbce pasty oznaczona zgodnie z tą metodą jest wyrażona w procentach masowych.
3. ZASADA
Oznaczenie jest wykonywane metodą chromatografii gazowej. Fluor ze związków zawierających fluor zostaje przekształcony w trietylofluorosilan (TEFS) w bezpośredniej reakcji z chlorotrietylosilanem (TECS) w kwaśnym roztworze i równocześnie ekstrahowany ksylenem zawierającym cykloheksan jako wewnętrzny standard.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. Fluorek sodu wysuszony w temperaturze 120°C do stałej masy.
4.2. Woda redestylowana lub o równoważnej jakości.
4.3. Kwas solny, d
4.4. Cykloheksan.
4.5. Ksylen, który nie daje żadnych pików przed pikiem tego rozpuszczalnika wówczas, kiedy jest analizowany chromatograficznie w tych samych warunkach co próbka (6.1). Jeżeli niezbędne, jest oczyszczany przez destylację (5.8).
4.6. Chlorotrietylosilan (TECS Merck lub równoważny).
4.7. Roztwory standardowe fluoru
4.7.1. Roztwór bazowy, 0,250 mg F-/ml. Odważyć dokładnie 138,1 mg fluorku sodu (4.1) i rozpuścić w wodzie (4.2). Przenieść ilościowo roztwór do 250 ml kolby miarowej (5.5). Rozcieńczyć do kreski wodą (4.2) i wymieszać.
4.7.2. Rozcieńczony roztwór bazowy 0,050 mg F-/1 ml. Przenieść pipetą 20 ml roztworu bazowego (4.7.1) do 100 ml kolby miarowej (5.5). Rozcieńczyć do kreski wodą i wymieszać.
4.8. Roztwór wewnętrznego standardu
Zmieszać 1 ml cykloheksanu (4.4) i 5 ml ksylenu (4.5).
4.9. Chlorotrietylosilan / roztwór wewnętrznego standardu
Przenieść pipetą (5.7) 0,6 ml chlorotrietylosilanu (4.6) i 0,12 ml roztworu wewnętrznego standardu (4.8) do 10 ml kolby miarowej. Rozcieńczyć ksylenem (4.5) do kreski i wymieszać. Przygotować świeżo tego samego dnia.
4.10. Kwas nadchlorowy, 70% (m/v).
4.11. Kwas nadchlorowy, 20% (m/v) w wodzie (4.2).
5. APARATURA
5.1. Zwykły sprzęt laboratoryjny.
5.2. Chromatograf gazowy wyposażony w detektor płomieniowo-jonizacyjny.
5.3. Obrotowy mikser Vortex lub równoważny.
5.4. Wytrząsarka Bühler typ SMB 1 lub równoważna.
5.5. Kolby miarowe, 100 i 250 ml, wykonane z polipropylenu.
5.6. Probówki wirówkowe (szklane); 20 ml z korkiem gwintowanym pokrytym teflonem, Sovirel typ 611–56 lub równoważne. Czyścić probówki i korki gwintowane przez ługowanie w ciągu kilkunastu godzin w kwasie nadchlorowym (4.11) i następnie pięciokrotne kolejne płukanie wodą (4.2) i końcowe suszenie w temperaturze 100°C.
5.7. Pipety, odpowiednie do odmierzania objętości od 50 do 200 µl, z jednorazowymi plastikowymi zakończeniami.
5.8. Aparatura do destylacji, wyposażona w kolumnę Schneidera z trzema kulami lub równoważna kolumna Vigreux.
6. PROCEDURA
6.1. Analiza próbki
6.1.1. Wybrać tubę pasty do zębów uprzednio nieotwieraną, obciąć dolną część tuby i usunąć całą zawartość do plastikowego pojemnika, wymieszać starannie i przechowywać w warunkach zapobiegających pogorszeniu jakości.
6.1.2. Odważyć dokładnie 150 mg (m) próbki do probówki wirówkowej (5.6), dodać 5 ml wody (4.2) i homogenizować (5.3).
6.1.3. Dodać 1 ml ksylenu (4.5).
6.1.4. Dodawać kroplami 5 ml kwasu solnego (4.3) i homogenizować (5.3).
6.1.5. Dodać pipetą 0,5 ml roztworu chlorotrietylosilanu / standardu wewnętrznego (4.9) do probówki wirówkowej (5.6).
6.1.6. Zamknąć probówkę gwintowanym korkiem (5.6) i mieszać dokładnie w ciągu 45 minut na wytrząsarce (5.4) ustawionej na 150 wstrząsów na minutę.
6.1.7. Wirować 10 minut z taką szybkością, aby otrzymać wyraźny rozdział warstw, otworzyć probówkę, odciągnąć warstwę organiczną i wprowadzić strzykawką 3 µl warstwy organicznej na kolumnę chromatografu gazowego (5.2).
Uwaga: Eluowanie wszystkich składników trwa ok. 20 minut.
6.1.8. Powtórzyć analizę, obliczyć stosunki średnich powierzchni pików (Atefs/Ach) i odczytać odpowiednie ilości fluoru (w miligramach m1) z wykresu kalibracyjnego (6.3).
6.1.9. Obliczyć całkowitą zawartość fluoru w próbce (w procentach masowych fluoru), jak wskazano w pkt 7.
6.2. Warunki analizy chromatograficznej
Kolumna: stal nierdzewna
Długość: 1,8 m
Średnica: 3 mm
Nośnik: Gaschrom Q 80–100 mesh
Faza stacjonarna: olej silikonowy DC 200 lub równoważny, 20%, kondycjonowanie kolumny przez noc w temperaturze 100°C (gaz nośny azot, przepływ 25 ml na minutę) i powtarzane każdej nocy. Po każdych czterech lub pięciu wstrzyknięciach ponownie kondycjonować kolumnę przez ogrzewanie w temperaturze 100°C w ciągu 30 minut.
Temperatura:
kolumna: 70°C,
dozownik: 150°C,
detektor: 250°C.
Przepływ gazu nośnego: 35 ml azotu na minutę.
6.3. Wykres kalibracyjny
6.3.1. Umieścić pipetą w serii sześciu probówek wirówki (5.6) po 0, 1, 2, 3, 4 i 5 ml rozcieńczonego standardowego roztworu fluoru (4.7.2). Uzupełnić objętość w każdej probówce wodą do 5 ml (4.2).
6.3.2. Postępować tak, jak opisano w pkt 6.1.3–6.1.6 włącznie.
6.3.3. Wprowadzić strzykawką 3 µl warstwy organicznej na kolumnę chromatografu gazowego (5.2).
6.3.4. Powtórzyć wstrzyknięcie i obliczyć średni stosunek pików (Atefs/Ach).
6.3.5. Narysować wykres kalibracyjny współzależności masy fluoru (w miligramach) w roztworach standardowych (6.3.1) i stosunku powierzchni pików (Atefs/Ach) mierzonych zgodnie z pkt 6.3.4. Połączyć punkty wykresu najbardziej odpowiednią linią prostą obliczoną równaniem regresji.
7. OBLICZENIE
Stężenie całkowitej zawartości fluoru w próbce (w procentach masowych fluoru) (% m/m F) jest określone zgodnie z równaniem:
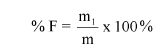
gdzie:
m = próbka analityczna (w miligramach) (6.1.2),
m1 = ilość F (w miligramach) odczytana z wykresu kalibracyjnego (6.1.8).
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości fluoru ok. 0,15% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,012% (m/m).
XXII. IDENTYFIKOWANIE I OZNACZANIE ORGANICZNYCH ZWIĄZKÓW RTĘCI
CEL I ZAKRES STOSOWANIA
Metoda może być stosowana do identyfikowania i oznaczania organicznych pochodnych rtęci stosowanych jako środki konserwujące w produktach kosmetycznych używanych w okolicach oczu. Mogą być stosowane do tiomersalu (INN) i fenylortęci i jej soli.
A. IDENTYFIKOWANIE
1. ZASADA
Organiczne związki rtęci są kompleksowane 1,5-difenylo-3-tiokarbazonem (ditizonem). Po ekstrakcji ditizonu czterochlorkiem węgla, jest wykonywana chromatografia cienkowarstwowa na żelu krzemionkowym. Plamy ditizonu pojawiają się w pomarańczowym kolorze.
2. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
2.1. Kwas siarkowy, 25% (v/v).
2.2. 1,5-difenylo-3-tiokarbazon(ditizon): 0,8 mg w 100 ml czterochlorku węgla (2.4).
2.3. Azot.
2.4. Czterochlorek węgla.
2.5. Rozpuszczalnik rozwijający: heksan/aceton, 90:10 (v/v).
2.6. Roztwór standardowy, 0,001% w wodzie:
2-(etylortęciotio)benzoesan sodu,
chlorek etylortęci lub chlorek metylortęci,
azotan fenylortęci lub octan fenylortęci,
chlorek rtęci (II) lub octan rtęci (II).
2.7. Przygotowane fabrycznie płytki z żelem krzemionkowym (np. Merck 5721 lub równoważne).
2.8. Chlorek sodu.
3. APARATURA
3.1. Zwykły sprzęt laboratoryjny.
3.2. Zwykła aparatura do chromatografii cienkowarstwowej.
3.3. Filtr rozdzielający fazy.
4. PROCEDURA
4.1. Ekstrakcja
4.1.1. Rozcieńczyć 1 g próbki w probówce wirówki przez dodawanie 20 ml wody destylowanej. Otrzymać jak największe rozproszenie i ogrzewać do temperatury 60°C w łaźni wodnej. Dodać 4 g chlorku sodu (2.8). Wytrząsnąć. Pozostawić do schłodzenia.
4.1.2. Wirować w ciągu co najmniej 20 minut przy 4500 obrotach/min w celu oddzielenia większej części substancji stałej z cieczy. Przesączyć do rozdzielacza i dodać 0,25 ml roztworu kwasu siarkowego (2.1).
4.1.3. Ekstrahować kilkakrotnie po 2 lub 3 ml roztworu ditizonu (2.2) do zaniku zielonego koloru ostatniej warstwy organicznej.
4.1.4. Przesączyć każdą warstwę organiczną przez filtr rozdzielający fazy (3.3).
4.1.5. Odparować do sucha w strumieniu azotu (2.3).
4.1.6. Rozpuścić w 0,5 ml czterochlorku węgla (2.4). Używać roztworu natychmiast do analizy, jak wskazano w pkt 4.2.1.
4.2. Rozdzielanie i identyfikowanie
4.2.1. Nanieść niezwłocznie 50 µl roztworu otrzymanego jak w opisie podanym w pkt 4.1.6 w czterochlorku węgla na płytkę z żelem krzemionkowym (2.7). Jednocześnie poddać 10 ml roztworu standardowego (2.6) takiemu postępowaniu, jak opisano w pkt 4.1, i nanieść 50 µl roztworu otrzymanego jak w opisie podanym w pkt 4.1.6 na tę samą płytkę.
4.2.2. Umieścić płytkę w rozpuszczalniku (2.5) i pozwolić mu wznosić się do wysokości 150 mm. Związki organiczne rtęci pojawiają się jako kolorowe plamy, których kolor jest trwały. Zabezpieczyć płytkę chromatograficzną szklaną płytką bezpośrednio po odparowaniu rozpuszczalnika.
Przykładowe otrzymane wartości Rf:
|
| Rf | Kolor |
| tiomersal | 0,33 | pomarańczowy |
| chlorek etylortęci | 0,29 | pomarańczowy |
| chlorek metylortęci | 0,29 | pomarańczowy |
| sole fenylortęci | 0,21 | pomarańczowy |
| sole rtęci | 0,10 | pomarańczowy |
| octan rtęci | 0,10 | pomarańczowy |
| 1,5-difenylo-3-tiokarbazon | 0,09 | różowy |
B. OZNACZANIE
1. DEFINICJA
Zawartość organicznych związków rtęci oznaczona według tej metody jest wyrażona jako procent masowy (m/m) rtęci w próbce.
2. ZASADA
Metoda polega na zmierzeniu ilości całej rtęci obecnej w próbce. Konieczne jest zatem w pierwszej kolejności upewnienie się, że w próbce nie występuje rtęć w postaci nieorganicznej, aby móc identyfikować organiczne pochodne rtęci zawarte w próbce wyrobu. Po mineralizacji uwolniona rtęć jest oznaczana przez bezpłomieniową absorpcję atomową.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. Stężony kwas azotowy d
3.2. Stężony kwas siarkowy d
3.3. Woda redestylowana.
3.4. Nadmanganian potasu, 7% (m/v) roztwór.
3.5. Chlorowodorek hydroksyloamonowy, 1,5% (m/v) roztwór.
3.6. Nadsiarczan potasu, 5% (m/v) roztwór.
3.7. Chlorek cyny (II) (chlorek cynawy), 10% (m/v) roztwór.
3.8. Stężony kwas solny, d
3.9. Wata szklana impregnowana chlorkiem palladu (II) (chlorkiem palladowym), 1% (m/m).
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Aparatura do oznaczania rtęci metodą bezpłomieniowej absorpcji atomowej (technika zimnej pary) włącznie z niezbędnym sprzętem szklanym. Droga optyczna naczynka pomiarowego powinna wynosić co najmniej 100 mm.
5. PROCEDURA
Przedsięwziąć zwykłe środki ostrożności przy śladowej analizie rtęci.
5.1. Rozkład próbki
5.1.1. Odważyć dokładnie 150 mg próbki (m). Dodać 10 ml kwasu azotowego (3.1) i pozostawić próbkę jego działaniu na trzy godziny w kolbie hermetycznej w łaźni wodnej o temperaturze 55°C, wstrząsając w regularnych odstępach czasu. W tym samym czasie wykonać ślepą próbę z samymi odczynnikami.
5.1.2. Po schłodzeniu dodać 10 ml kwasu siarkowego (3.2) i ponownie umieścić w łaźni wodnej o temperaturze 55°C na 30 minut.
5.1.3. Umieścić kolbę w łaźni lodowej i dodać ostrożnie 20 ml wody (3.3).
5.1.4. Dodawać porcjami po 2 ml 7% roztwór nadmanganianu (VII) potasu aż do odbarwienia roztworu. Ponownie umieścić w łaźni wodnej o temperaturze 55°C na następne 15 minut.
5.1.5. Dodać 4 ml roztworu nadsiarczanu (VI) potasu (3.6). Kontynuować ogrzewanie w łaźni wodnej o temperaturze 55°C przez 30 minut.
5.1.6. Pozostawić do schłodzenia i przenieść zawartość kolby do 100 ml kolby miarowej. Popłukać kolbę 5 ml chlorowodorku hydroksyloamonowego (3.5) i następnie płukać czterokrotnie porcjami po 10 ml wody (3.3). Roztwór powinien być zupełnie bezbarwny. Uzupełnić wodą (3.3) do kreski.
5.2. Oznaczanie
5.2.1. Umieścić 10 ml badanego roztworu (5.1.6) w szklanym naczyniu aparatury do oznaczania rtęci techniką zimnej pary (4.2). Rozpuścić w 100 ml wody (3.3), a następnie dodać 5 ml kwasu siarkowego (VT) (3.2) i 5 ml roztworu chlorku cyny (II) (3.7). Wymieszać po każdym dodatku. Poczekać 30 sekund do zredukowania w całości jonów rtęci do stanu metalicznego i odczytać wartość (n).
5.2.2. Umieścić watę szklaną impregnowaną chlorkiem palladu (II) między naczyniem do redukowania rtęci i naczynkiem przepływowym aparatury (4.2). Powtórzyć postępowanie opisane w pkt 5.2.1 i zarejestrować odczyt. Jeżeli różni się on od zera, oznacza to, że mineralizacja nie zaszła całkowicie i analizę należy powtórzyć.
6. OBLICZENIE
Przyjęto:
m = masa (w miligramach) próbki analitycznej,
n = ilość rtęci (w µg) odczytana na skali instrumentu.
Ilość rtęci wyrażona jako procent masowy rtęci, jest obliczana zgodnie z wzorem:

7. UWAGI
7.1. Dla poprawienia mineralizacji może być konieczne rozpoczęcie postępowania od rozcieńczenia roztworu.
7.2. Jeżeli spodziewana jest absorpcja rtęci przez substrat, ilościowe oznaczanie powinno być wykonane metodą dodawania standardu.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
W przypadku stężenia rtęci 0,007% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć wartości absolutnej 0,00035%.
XXIII. OZNACZANIE SIARCZKÓW METALI ALKALICZNYCH I ZIEM ALKALICZNYCH
1. CEL I ZAKRES STOSOWANIA
Metoda opisuje oznaczanie siarczków obecnych w produktach kosmetycznych. Obecność tioli lub innych środków redukujących (łącznie z siarczynami) (IV) nie przeszkadza w oznaczeniu.
2. DEFINICJA
Stężenie siarczków oznaczone tą metodą jest wyrażone jako procent masowy siarki.
3. ZASADA
Po zakwaszeniu środowiska siarkowodór jest unoszony przez strumień azotu i wiązany w postaci siarczku kadmowego. Ten ostatni jest odsączony i przemyty, a następnie oznaczony jodometrycznie.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. Stężony kwas solny, d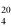
4.2. Tiosiarczan (VI) sodu, 0,1 M standardowy roztwór.
4.3. Jod, 0,05 M standardowy roztwór.
4.4. Siarczek sodu.
4.5. Octan kadmu (II).
4.6. Stężony amoniak, d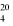
4.7. Amoniakalny roztwór octanu kadmu (II): rozpuścić 10 g octanu kadmu (II) (4.5) w ok. 50 ml wody. Dodać amoniak (4.6) aż do ponownego rozpuszczenia osadu (średnio ok. 20 ml). Uzupełnić wodą do kreski 100 ml.
4.8. Azot.
4.9. Roztwór amoniaku M.
5. APARATURA
5.1. Zwykły sprzęt laboratoryjny.
5.2. 100 ml kolba okrągłodenna z trzema szyjami ze szlifem.
5.3. Dwie 150 ml kolby stożkowe ze szlifem wyposażone w urządzenia obejmujące rurkę zanurzeniową i boczną rurkę wylotową do wypuszczenia powstającego gazu.
5.4. Lejek do sączenia z długą nóżką.
6. PROCEDURA
6.1. Wprowadzenie siarczków.
6.1.1. Pobrać opakowanie, którego poprzednio nie otwierano. Odważyć dokładnie w kolbie okrągłodennej (5.2) masę (m) (wyrażoną w gramach) wyrobu odpowiadającą nie więcej niż 30 mg jonów siarczkowych. Dodać 60 ml wody i dwie krople cieczy przeciwdziałającej pienieniu.
6.1.2. Przenieść po 50 ml roztworu (4.7) do każdej z dwóch kolb stożkowych (5.3).
6.1.3. Umieścić wkraplacz, rurkę zanurzeniową i rurkę wylotową w kolbie okrągłodennej (5.2). Przyłączyć rurkę wylotową do serii kolb stożkowych (5.3) przewodami z PCV. Uwaga: Aparatura wprowadzająca powinna przejść następujący test szczelności. Symulując warunki testu, zastąpić próbkę wyrobu przeznaczoną do wykonania oznaczenia przez 10 ml roztworu siarczku (przygotowanego jak w opisie w pkt 4.4) zawierającego „X mg” siarczku (oznaczonego jodometrycznie). Przyjęto „Y” jako liczbę miligramów siarczku stwierdzoną w końcu tej operacji. Różnica między ilością „X” i ilością „Y” nie powinna przekroczyć 3%.
6.1.4. Przepuszczać azot (4.8) przez 15 minut, z szybkością dwóch pęcherzyków gazowych na sekundę, w celu usunięcia powietrza zawartego w kolbie okrągłodennej (5.2).
6.1.5. Ogrzewać kolbę okrągłodenną do tempertatury 85 +/– 5°C.
6.1.6. Zatrzymać strumień azotu (4.8) i dodać 40 ml kwasu solnego (4.1), kropla po kropli.
6.1.7. Wprowadzić ponownie strumień azotu (4.8) wtedy, kiedy prawie cały kwas został wkroplony, pozostawiając minimalną ciekłą warstwę (uszczelnienie), aby zapobiec wyciekom siarkowodoru.
6.1.8. Przerwać ogrzewanie po 30 minutach. Pozostawić kolbę (5.2) do schłodzenia i kontynuować przepływ azotu (4.8) przez co najmniej półtorej godziny.
6.2. Miareczkowanie
6.2.1. Przesączyć siarczek kadmu przez lejek z długą nóżką (5.4).
6.2.2. Popłukać kolby stożkowe (5.3) najpierw roztworem amoniaku (4.9) i przelać go przez lejek. Następnie popłukać wodą destylowaną i użyć wody do przemycia osadu zatrzymanego przez filtr.
6.2.3. Przemyć osad 100 ml wody.
6.2.4. Umieścić bibułę filtracyjną z lejka zawierającą osad w pierwszej kolbie stożkowej. Dodać 25 ml (n) roztworu jodu (4.3), ok. 20 ml kwasu solnego (4.1) i 50 ml wody destylowanej.
6.2.5. Oznaczyć nadmiar jodu roztworem tiosiarczanu (VI) sodu (n2) (4.2).
7. OBLICZENIE
Zawartość siarczku w próbce, wyrażona jako procent masowy siarki, jest obliczana według wzoru:
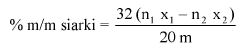
gdzie:
n1 = ilość (w mililitrach) zużytego standardowego roztworu jodu (4.3),
x1 = stężenie molowe tego roztworu,
n2 = ilość (w mililitrach) standardowego roztworu tiosiarczanu (VI) sodu (4.2),
x2 = stężenie molowe tego roztworu,
m = masa (w gramach) próbki analitycznej.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości siarczku ok. 2% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć wartości absolutnej 0,2% (m/m).
XXIV. IDENTYFIKOWANIE I OZNACZANIE 1-(4-AMINOBENZOILO) GLICEROLU
A. IDENTYFIKOWANIE
1. PRZEDMIOT I ZAKRES ZASTOSOWANIA
Metodę stosuje się do wykrywania 1-(4-aminobenzoilo) glicerolu (INCI: glycerylpaba). Można tą metodą wykrywać również 4-aminobenzoesan etylu (INN: benzocaine), który może występować jako zanieczyszczenie.
2. ZASADA
Identyfikowanie przeprowadza się metodą chromatografii cienkowarstwowej na żelu krzemionkowym ze wskaźnikiem fluorescencyjnym przez wykrywanie wolnej pierwszorzędowej grupy aminowej i pojawienie się plamy barwnika dwufazowego na płytce.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. Mieszanina rozpuszczalników: cykloheksan / propan-2-ol / stabilizowany dichlorometan (48:64:9 obj.).
3.2. Rozpuszczalnik rozwijający: eter naftowy (40–60) / benzen / aceton / roztwór wodorotlenku amonu (co najmniej 25% NH3,) (35:35:35:1 obj.).
3.3. Roztwór wywołujący
1) azotan (III) sodu: 1g w 100 ml i 1 M kwasu chlorowodorowego (przygotowany bezpośrednio przed użyciem);
2) 2-naftol: 0,2 g w 100 ml 1 M wodorotlenku potasu.
3.4. Roztwory standardowe
1-(4-aminobenzoilo) glicerolu: 0,05 g w 100 ml mieszaniny rozpuszczalników (3.1),
4-aminobenzoesan etylu: 0,05 g w 100 ml mieszaniny rozpuszczalników (3.1).
3.5. Płytki z żelem krzemionkowym 60 F254, grubość 0,25 mm, 200 x 200 mm.
4. APARATURA
4.1. Zwykła aparatura do chromatografii cienkowarstwowej.
4.2. Wibrator ultradźwiękowy.
4.3. Filtr z wkładem mikroporowatym F H 0,5 µm lub równoważny.
5. PROCEDURA
5.1. Przygotowanie próbki
W 10 ml kolbie miarowej z korkiem zważyć 1,5 g analizowanego wyrobu. Uzupełnić rozpuszczalnikiem (3.1) do kreski. Zamknąć i pozostawić na okres jednej godziny w temperaturze pokojowej w łaźni ultradźwiękowej (4.2). Przesączyć przez filtr Milipore (4.3) i używać filtratu do analizy chromatograficznej.
5.2. Chromatografia cienkowarstwowa
Nanieść po 10 µl roztworu próbki (5.1) i wszystkich roztworów standardowych (3.4) na płytkę (3.5).
Rozwijać chromatogram do wysokości 150 mm w komorze uprzednio wysycanej rozpuszczalnikiem (3.2). Pozostawić płytkę do wysuszenia w temperaturze otoczenia.
5.3. Rozwijanie
5.3.1. Obserwować płytkę w świetle UV przy długości fali 254 nm.
5.3.2. Spryskać całkowicie wysuszoną płytkę roztworem (3.3 ppkt 1).
Pozostawić do wysuszenia w temperaturze otoczenia w ciągu 1 minuty i niezwłocznie spryskać roztworem (3.3 ppkt 2).
Wysuszyć płytkę w suszarce w temperaturze 60°C. Plamy aminobenzoesanów pojawiają się w kolorze pomarańczowym. Czas retencji: 1-(4-aminobenzoilo) glicerolu Rf = 0,07; 4-aminobenzoesanu etylu Rf = 0,55.
B. OZNACZANIE
1. PRZEDMIOT I ZAKRES
W tej metodzie oznacza się 1-(4-aminobenzoilo) glicerol. Może ona również być stosowana do oznaczania 4-aminobenzoesanu etylu. Nie można oznaczyć większego stężenia niż 5% masowych 1-(4-aminobenzoilo) glicerolu i 1% masowy 4-aminobenzoesanu etylu.
2. DEFINICJA
Zawartości 1-(4-aminobenzoilo) glicerolu i 4-aminobenzoesanu etylu oznaczane tą metodą są wyrażane w procentach masowych (% m/m) wyrobu.
3. ZASADA
Analizowany wyrób jest zawieszony w metanolu i po odpowiedniej obróbce próbki oznaczenie jest prowadzone metodą wysokosprawnej chromatografii cieczowej (HPLC).
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy, a tam gdzie to konieczne – odpowiadać HPLC.
4.1. Metanol.
4.2. Diwodoroortofosforan (V) potasu (KH2PO4).
4.3. 2-hydrat octanu cynku (II) (Zn(CH3COO)2 2H20.
4.4. Kwas octowy, d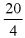
4.5. 3'hydrat heksacyjanożelazianu (II) (żelazocyjanku) tetrapotasu (K4(Fe(CN)6) 3H2O).
4.6. 4-hydroksybenzoesan etylu.
4.7. 1-(4-aminobenzoesan) glicerolu.
4.8. 4-aminobenzoesan etylu.
4.9. Roztwór buforu fosforanowego (0,02 M): rozpuścić 2,72 g diwodoroortofosforanu (V) potasu (4.2) w jednym litrze wody.
4.10. Roztwór wymywający: roztwór buforu fosforanowego (4.9) / metanol (4.1) (61:39 obj.).
Skład fazy ruchomej można zmieniać w celu uzyskania współczynnika rozdziału R ≥ 1,5 obliczonego według wzoru:
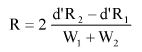
gdzie:
R1 i R2 = czas retencji pików, w minutach,
W1 i W2 = szerokość pików w połowie wysokości, w milimetrach.
d' = szybkość przesuwu papieru, w milimetrach na minutę.
4.11. Bazowy roztwór 1-(4-aminobenzoesanu) glicerolu: zważyć dokładnie ok. 40 mg 1-(4-aminobenzoilo) glicerolu i umieścić w 100 ml kolbie miarowej. Rozpuścić w 40 ml metanolu (4.1). Uzupełnić do kreski roztworem buforowym (4.9) i wymieszać.
4.12. Bazowy roztwór 4-aminobenzoesanu etylu: zważyć dokładnie ok. 40 mg 4-aminobenzoesanu etylu i umieścić w 100 ml kolbie miarowej, rozpuścić w 40 ml metanolu (4.1), uzupełnić do kreski roztworem buforowym i wymieszać.
4.13. Roztwór wewnętrznego standardu: zważyć dokładnie ok. 50 mg 4-hydroksybenzoesanu etylu (4.6), przenieść do 100 ml kolby miarowej, rozpuścić w 40 ml metanolu (4.1), uzupełnić do kreski roztworem buforowym (4.9) i wymieszać.
4.14. Roztwory standardowe: przygotować cztery roztwory standardowe przez rozpuszczenie w 100 ml eluentu (4.10) odpowiednich związków zgodnie z następującą tabelą:
| Roztwór standardowy | 1-(4-aminobenzoesan) glicerolu | 4-aminobenzoesan etylu | 4-hydroksybenzoesan etylu | |||
| (µg/ml)* | ml (4.11) | (µg/ml)* | ml (4.12) | (µg/ml)* | ml (4.13) | |
| I II III IV | 8 16 24 40 | 2 4 6 10 | 8 12 16 20 | 2 3 4 5 | 50 50 50 50 | 10 10 10 10 |
| * Powyższe wartości wskazano jako orientacyjne, a odpowiadają one dokładnie masom podanym w pkt 4.11, 4.12 i 4.13. | ||||||
4.15. I roztwór Carreza: rozpuścić 26,5 g heksacyjanożelazianu (III) tetrapotasu (4.5) w wodzie i uzupełnić do 250 ml.
4.16. II roztwór Carreza: rozpuścić 54,9 octanu cynku (II) (4.3) i 7,5 ml kwasu octowego (4.4) w wodzie i uzupełnić do 250 ml.
4.17. Wypełnienie Merck Lichrosorb RP-18 lub równoważne o średnim rozmiarze cząstek 5 µm.
5. APARATURA
5.1. Zwykłe wyposażenie laboratoryjne.
5.2. Wyposażenie do wysokosprawnej chromatografii cieczowej z detektorem UV ze zmienną długością fali i zestaw komór termostatowany w temperaturze 45°C.
5.3. Kolumna ze stali nierdzewnej, długość: 250 mm, średnica wewnętrzna: 4,6 mm; wypełnienie: Lichrosorb RP-18 (4.17).
5.4. Łaźnia ultradźwiękowa.
6. PROCEDURA
6.1. Przygotowanie próbki
6.1.1. W 100 ml zlewce zważyć dokładnie ok. 1 g próbki i dodać 10 ml metanolu (4.1).
6.1.2. Umieścić zlewkę w łaźni ultradźwiękowej (5.4) na 20 minut w celu otrzymania suspensji. Przenieść ilościowo otrzymaną suspensję do 100 ml kolby miarowej, w której znajduje się nie więcej niż 75 ml roztworu wymywającego (4.10).
Dodać kolejno po 1 ml roztworu Carreza (4.15 i 4.16) i wymieszać po każdym dodaniu. Uzupełnić do kreski eluentem (4.10), ponownie wymieszać i przesączyć przez karbowany sączek bibułowy.
6.1.3. Przenieść pipetą 3,0 ml przesączu otrzymanego jak w opisie pkt 6.1.2 i 5,0 ml roztworu standardu wewnętrznego (4.13) do 50 ml kolby miarowej. Uzupełnić do kreski roztworem wymywającym (4.10) i wymieszać. Stosować tak otrzymany roztwór do wykonania analizy chromatograficznej opisanej w pkt 6.2.
6.2. Chromatografia
6.2.1. Ustawić przepływ fazy ruchomej (4.10) na 1,2 ml/min, a temperaturę kolumny na 45oC.
6.2.2. Ustawić detektor (5.2) na długość fali 274 nm.
6.2.3. Wprowadzić mikrostrzykawką co najmniej dwukrotnie po 20 µl roztworu (6.1.3) do chromatografu i zmierzyć powierzchnię pików.
6.3. Krzywa odwzorowania
6.3.1. Wprowadzić kolejno po 20 µl wszystkich roztworów standardowych (4.14) i zmierzyć powierzchnię pików.
6.3.2. Dla każdego stężenia obliczyć stosunek między powierzchniami piku 1-(4-aminobenzoesanu) glicerolu i powierzchniami piku wewnętrznego standardu. Nanieść ten stosunek na osi odciętych, a na osi rzędnych stosunek odpowiednich mas.
6.3.3. W ten sam sposób postępować dla 4-hydroksybenzoesanu etylu. 7. OBLICZENIA
7.1. Z krzywej odwzorowania otrzymanej zgodnie z pkt 6.3 odczytać stosunki mas (RP1, RP2) odpowiadające stosunkom między powierzchniami pików obliczonymi zgodnie z pkt 6.2.3, gdzie:
RP1 = stosunek: masa 1-(4-aminobenzoesanu)glicerolu / masa 4-hydroksybenzoesanu etylu.
RP2 = stosunek: masa 4-aminobenzoesanu etylu / masa 4-hydroksybenzoesanu etylu.
7.2. Ze stosunków mas otrzymanych w ten sposób obliczyć zawartość 1-(4-aminobenzoesanu) glicerolu i 4-aminobenzoesanu etylu jako procenty masowe (% m/m) według wzorów:
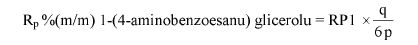

q = ilość 4-hydroksybenzoesanu etylu (wewnętrznego standardu), w miligramach (4.12),
p = ilość próbki, w gramach (6.1.1).
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
8.1. Dla zawartości 5% (m/m) 1-(4-aminobenzoesanu) glicerolu różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekroczyć 0,25%.
8.2. Dla zawartości 1% (m/m) 4-aminobenzoesanu etylu różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekroczyć 0,10%.
9. UWAGI
9.1. Przed wykonaniem analizy należy sprawdzić, czy próbka nie zawiera substancji, których piki mogłyby się pokrywać częściowo z pikiem wewnętrznego standardu (4-hydroksybenzoesanu etylu) na chromatogramie.
9.2. Aby sprawdzić, czy nie ma zakłóceń, należy powtórzyć oznaczenie, zmieniając proporcje metanolu w fazie ruchomej odpowiednio o 10%.
XXV. OZNACZANIE CHLOROBUTANOLU
1. PRZEDMIOT I ZAKRES ZASTOSOWANIA
Metoda jest odpowiednia do oznaczania chlorobutanolu (INCI: chlorobutanol) w stężeniu maksymalnym do 0,5% (m/m) w każdym produkcie kosmetycznym z wyjątkiem aerozoli.
2. DEFINICJA
Zawartość chlorobutanolu zmierzona tą metodą jest wyrażona w procentach masowych (% m/m) wyrobu.
3. ZASADA
Po odpowiednim przygotowaniu analizowanego wyrobu oznaczenie wykonuje się metodą chromatografii gazowej, stosując 2,2,2,-trichloroetanol jako wewnętrzny standard.
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
4.1. Chlorobutanol (1,1,1-trichloro-2-metylopropan-2-ol).
4.2. 2,2,2-trichloroetanol.
4.3. Etanol absolutny.
4.4. Roztwór standardowy chlorobutanolu: 0,025 g w 100 ml etanolu (4.3) (m/v).
4.5. Roztwór standardowy 2,2,2-trichloroetanolu: 4 mg w 100 ml etanolu (4.3) (m/v).
5. APARATURA
5.1. Zwykłe wyposażenie laboratoryjne.
5.2. Chromatograf gazowy z detektorem wychwytowym, Ni 63.
6. PROCEDURA
6.1. Przygotowanie próbki
Zważyć dokładnie między 0,1 i 0,3 g próbki. Umieścić odważkę w 100 ml kolbie miarowej. Rozpuścić w etanolu (4.3), dodać 1 ml roztworu standardu wewnętrznego (4.5) i uzupełnić do kreski etanolem (4.3).
6.2. Warunki chromatografii gazowej
6.2.1. Warunki działania muszą zapewnić współczynnik rozdziału R ≥ 1,5 obliczony według wzoru:
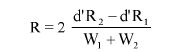
gdzie:
R1 i R2 = czas retencji pików, w minutach,
W1 i W2 = szerokość pików w połowie wysokości, w milimetrach,
d' = szybkość przesuwu papieru, w milimetrach na minutę.
6.2.2. Wymagany rozdział otrzymano w podanych poniżej warunkach:
| Kolumna |
| I | II |
| Materiał |
| szkło | stal nierdzewna |
| Długość |
| 1,80 m | 3 m |
| Średnica |
| 3 mm | 3 mm |
| Faza spoczynkowa |
| 10% Carbowax 20 M TPA na Gaschrom Q 80–100 mesh | 5% OV–17 na Chromosorb WAW DMCS 80–100 mesh |
| Konfekcjonowanie |
| 2 do 3 dni w temperaturze 190° C |
|
| Temperatura: |
|
|
|
|
| dozownik | 200°C | 150°C |
|
| kolumna | 150°C | 100°C |
|
| detektor | 200°C | 150°C |
| Gaz nośny |
| azot | argon / metan (95/5 v/v) |
| Przepływ |
| 35 ml/min | 35 ml/min |
6.3. Krzywa standardowa
W pięciu 100 ml kolbach miarowych umieścić kolejno po 1 ml roztworu standardowego (4.5) i po 0,2; 0,3; 0,4; 0,5 i 0,6 ml roztworu standardowego (4.4), uzupełnić do kreski etanolem (4.3) i wymieszać. Wprowadzić strzykawką po 1 µl każdego z tych roztworów do chromatografu zgodnie z warunkami działania podanymi w pkt 6.2.2 i narysować krzywą odwzorowania, nanosząc na osi odciętych stosunek masy chlorobutanolu do masy 2,2,2-trichloroetanolu, a na osi rzędnych stosunek odpowiednich powierzchni pików.
6.4. Wprowadzić strzykawką 1 µl roztworu otrzymanego w sposób opisany w pkt 6.1 i postępować zgodnie z warunkami opisanymi w pkt 6.2.2.
7. OBLICZENIA
7.1. Z krzywej standardowej (6.3) obliczyć ilość „a” wyrażoną jako µg chlorobutanolu w roztworze (6.1).
7.2. Zawartość chlorobutanolu w próbce oblicza się zgodnie z wzorem:
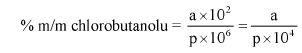
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości chlorobutanolu 0,5% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,01%.
Uwaga: Jeżeli wynik jest równy maksymalnemu dopuszczalnemu stężeniu lub je przekracza, należy sprawdzić, czy nie występuje współoznaczanie składników.
XXVI. IDENTYFIKOWANIE I OZNACZANIE CHININY
A. IDENTYFIKOWANIE
1. PRZEDMIOT I ZAKRES ZASTOSOWANIA
Metoda jest przeznaczona do wykrywania obecności chininy (INCI: quinine) w szamponach i płynach do włosów.
2. ZASADA
Identyfikację przeprowadza się metodą chromatografii cienkowarstwowej na żelu krzemionkowym. Wykrywanie chininy polega na stwierdzeniu fluorescencji w kwaśnym środowisku przy 360 nm.
Dla dalszego potwierdzenia fluorescencję można eliminować, stosując opary bromu, a opary amoniaku spowodują pojawienie się żółtawej fluorescencji.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. Płytki z żelem krzemionkowym, bez wskaźników fluorescencji, o grubości 0,25 mm, 200 mm x 200 mm.
3.2. Rozpuszczalnik rozwijający: toluen / eter dietylowy / dichlorometan / dietyloamina: (20:20:20:8 obj.).
3.3. Metanol.
3.4. Kwas siarkowy (VI), 96%, d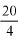
3.5. Eter dietylowy.
3.6. Środek rozwijający: dodać ostrożnie 5 ml kwasu siarkowego (VI) (3.4) do 95 ml eteru dietylowego (3.5) w chłodzonym pojemniku.
3.7. Brom.
3.8. Roztwór wodorotlenku amonu, 28%, d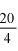
3.9. Chinina, bezwodna.
3.10. Roztwór standardowy: zważyć dokładnie ok. 100,0 mg bezwodnej chininy (3.9) w kolbie miarowej i rozpuścić w 100 ml metanolu (3.3).
4. APARATURA
4.1. Zwykłe wyposażenie do chromatografii cienkowarstwowej.
4.2. Łaźnia ultradźwiękowa.
4.3. Filtr miliporowy, FH 0,5 µm lub równoważny z odpowiednim wyposażeniem do sączenia.
5. PROCEDURA
5.1. Przygotowanie próbki
Zważyć dokładnie ilość próbki, która może zawierać w przybliżeniu 100 mg chininy w 100 ml kolbie miarowej, rozpuścić i uzupełnić do kreski metanolem (3.3). Zamknąć kolbę i zostawić na jedną godzinę w temperaturze otoczenia w łaźni ultradźwiękowej (4.2). Przesączyć (4.3) i stosować przesącz do analizy chromatograficznej.
5.2. Chromatografia cienkowarstwowa
Nanieść po 1,0 µl roztworu standardowego (3.10) i 1,0 µl roztworu próbki (5.1) na płytkę z żelem krzemionkowym (3.1). Rozwijać chromatogram na wysokość 150 mm z użyciem rozpuszczalnika (3.2) w komorze wysycanej uprzednio rozpuszczalnikiem (3.2).
5.3. Rozwijanie
5.3.1. Suszyć płytkę w temperaturze otoczenia.
5.3.2. Spryskać płytkę odczynnikiem (3.6).
5.3.3. Pozostawić płytkę do wysuszenia w ciągu jednej godziny w temperaturze otoczenia.
5.3.4. Obserwować płytkę w świetle lampy UV nastawionej na długość fali 360 nm. Chinina pojawia się jako plama o intensywnej błękitnej fluorescencji.
W tablicy poniżej podano przykładowe wartości retencji Rf głównych alkaloidów w porównaniu z czasem retencji chininy przy rozwijaniu chromatogramu rozpuszczalnikiem (3.2).
| Alkaloid | Rf |
| chinina | 0,20 |
| chinidyna | 0,29 |
| cynchonina | 0,33 |
| cynchonidyna | 0,27 |
| hydrochinidyna | 0,17 |
5.3.5. Dla dalszego potwierdzenia obecności chininy płytkę poddać przez okres ok. jednej godziny działaniu par bromu (3.7). Fluorescencja znika. Kiedy ta sama płytka jest poddana działaniu par amoniaku (3.8), pojawiają się ponownie plamy koloru brązowego, a kiedy ponownie bada się płytkę w świetle UV przy 360 nm, można zaobserwować żółtawą fluorescencję. Czułość (granica) identyfikacji: 0,1 µg chininy.
B. OZNACZANIE
1. PRZEDMIOT I ZAKRES ZASTOSOWANIA
Metoda opisuje oznaczanie chininy. Może być zastosowana do oznaczania maksymalnego dopuszczonego stężenia 0,5% (m/m) w szamponach i 0,2% (m/m) w płynach do włosów.
2. DEFINICJA
Zawartość chininy jest wyrażona w procentach masowych (% m/m) wyrobu.
3. ZASADA
Po odpowiednim przygotowaniu analizowanego produktu oznaczanie wykonuje się metodą wysokosprawnej chromatografii cieczowej (HPLC).
4. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy i odpowiednie do stosowania w HPLC.
4.1. Acetonitryl.
4.2. Diwodoroortofosforan (V) potasu (KH2PO4).
4.3. Kwas ortofosforowy (V), 85%, d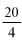
4.4. Bromek tetrametyloamonium.
4.5. Chinina, bezwodna.
4.6. Metanol.
4.7. Roztwór kwasu ortofosforowego (V) (0,1 M); zważyć 11,53 g kwasu ortofosforowego (V) (4.2) i rozpuścić w wodzie w 1000 ml kolbie miarowej.
4.8. Roztwór diwodoroortofosforanu (V) potasu (0,1 M); zważyć 13,6 g diwodoroortofosforanu (V) potasu (4.2) i rozpuścić w wodzie w 1000 ml kolbie miarowej.
4.9. Roztwór bromku tetrametyloamonium: rozpuścić 15,40 g bromku tetrametyloamonium (4.4) w wodzie w 1000 ml kolbie miarowej.
4.10. Roztwór wymywający: kwas ortofosforowy (V) (4.7) / diwodoroortofosforan (V) potasu (4.8) / bromek tetrametyloamonium (4.9) / woda / acetonitryl (4.1) (10:50:100:340:90 obj.). Skład fazy ruchomej można zmienić dla uzyskania współczynnika rozdziału R ≥ 1,5 według wzoru:
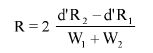
gdzie:
R1 i R2 = czas retencji pików, w minutach,
W1 i W2 = szerokość pików w połowie wysokości, w milimetrach,
d' = szybkość przesuwu papieru, w milimetrach na minutę.
4.11. Krzemionka poddana działaniu oktadecylosilanu, 10 µm.
4.12. Roztwory standardowe: zważyć dokładnie kolejno po ok. 5,0; 10,0; 15,0; i 20,0 mg bezwodnej chininy (4.5) w serii 100 ml kolb miarowych. Uzupełnić do kreski metanolem (4.6) i wytrząsać zawartość kolb do całkowitego rozpuszczenia chininy. Przesączyć każdą próbkę przez filtr 0,5 µm.
5. APARATURA
5.1. Zwykłe wyposażenie laboratoryjne.
5.2. Łaźnia ultradźwiękowa.
5.3. Wyposażenie do wysokosprawnej chromatografii cieczowej z detektorem o zmiennych długościach fali UV.
5.4. Kolumna: długość 250 mm, średnica wewnętrzna 4,6 mm, wypełnienie: krzemionka (4.11).
5.5. Miliporowy filtr FH 0,5 µm lub równoważny z odpowiednim zestawem do sączenia
6. PROCEDURA
6.1. Przygotowanie próbki
Zważyć dokładnie w 100 ml kolbie miarowej taką ilość wyrobu, aby zawierała 10,0 mg bezwodnej chininy, dodać 20 ml metanolu (4.6) i umieścić kolbę w łaźni ultradźwiękowej (5.2) na 20 minut. Uzupełnić do kreski metanolem (4.6). Wymieszać roztwór i następnie przesączyć jego część (5.5).
6.2. Chromatografia
Szybkość przepływu: 1,0 ml/min.
Długość fali w detektorze (5.3): 332 nm.
Objętość wprowadzanej próbki: 10 ml przesączonego roztworu (6.1).
Pomiar: powierzchnia piku.
6.3. Krzywa odwzorowania
Wprowadzić strzykawką co najmniej trzy razy po 10,0 µl każdego roztworu odniesienia (4.12), zmierzyć powierzchnię pików i obliczyć średnią powierzchnię dla każdego stężenia.
Przygotować krzywą odwzorowania i sprawdzić, czy jest prostoliniowa.
7. OBLICZENIA
7.1. Z krzywej odwzorowania (6.3) oznaczyć ilość bezwodnej chininy w µg obecnej we wprowadzonej objętości (6.2).
7.2. Stężenie bezwodnej chininy w próbce wyrażone jako procent masowy (% m/m) otrzymuje się według następującego wzoru:

gdzie:
B – ilość w mikrogramach bezwodnej chininy oznaczonej w 10 mikrolitrach przesączonego roztworu (6.1),
A – masa próbki w gramach (6.1).
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości bezwodnej chininy 0,5% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 0,02%. Dla zawartości bezwodnej chininy 0,2% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 0,01%.
XXVII. IDENTYFIKOWANIE I OZNACZANIE NIEORGANICZNYCH SIARCZANÓW I WODOROSIARCZANÓW
PRZEDMIOT I ZAKRES ZASTOSOWANIA
Metoda opisuje identyfikowanie i oznaczanie nieorganicznych siarczanów i wodorosiarczanów w produktach kosmetycznych. Jest odpowiednia tylko dla tych wyrobów, które zawierają fazę wodną lub alkoholową, i dla stężeń dwutlenku siarki nieprzekraczających 0,2%.
A. IDENTYFIKOWANIE
1. ZASADA
Próbkę ogrzewa się w kwasie solnym, a uwolniony dwutlenek siarki identyfikuje się zarówno na podstawie jego zapachu, jak i barwy papierka wskaźnikowego.
2. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
2.1. Kwas chlorowodorowy (4 M).
2.2. Papierek wskaźnikowy z jodkiem potasu i skrobią lub inny odpowiedni.
3. APARATURA
3.1. Zwykły sprzęt laboratoryjny.
3.2. Kolba (25 ml) z krótką chłodnicą zwrotną.
4. PROCEDURA
4.1. W kolbie (3.2) umieścić ok. 2,5 g próbki i 10 ml kwasu chlorowodorowego (2.1).
4.2. Wymieszać i ogrzewać do wrzenia.
4.3. Sprawdzić wydzielanie dwutlenku siarki zarówno przez ocenę zapachową, jak i papierem wskaźnikowym (2.2).
B. OZNACZANIE
1. DEFINICJA
Zawartość siarczanu (IV) lub wodorosiarczanu (IV) oznaczona tą metodą jest wyrażona jako procent masowy dwutlenku siarki.
2. ZASADA
Po zakwaszeniu próbki uwolniony dwutlenek siarki destyluje się do roztworu nadtlenku wodoru. Powstały kwas siarkowy (VI) jest miareczkowany mianowanym roztworem wodorotlenku sodu.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. Nadtlenek wodoru 0,2% (m/v) przygotowany tego samego dnia.
3.2. Kwas ortofosforowy (V), d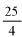
3.3. Metanol.
3.4. Wodorotlenek sodu (0,01 M), roztwór mianowany.
3.5. Azot.
3.6. Wskaźnik: mieszanina 1:1 (obj.) czerwieni metylowej (0,03% m/v w etanolu) i błękitu metylenowego 0,05% (m/v w etanolu). Przesączyć otrzymaną mieszaninę.
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Aparatura do destylacji (patrz: rys. 12).
5. PROCEDURA
5.1. Zważyć dokładnie ok. 2,5 g próbki do kolby destylacyjnej A (patrz: rys. 12).
5.2. Dodać 60 ml wody i 50 ml metanolu (3.3), wymieszać.
5.3. W odbieralniku destylatu D (patrz: rys. 12) umieścić 10 ml nadtlenku wodoru (3.1), 60 ml wody i kilka kropli wskaźnika (3.6). Dodać kilka kropli wodorotlenku sodu (3.4) do zmiany zabarwienia wskaźnika na kolor zielony.
5.4. Powtórzyć czynności opisane w pkt 5.3 dla płuczki do przemywania E (patrz: rys. 12).
5.5. Zmontować aparaturę i nastawić przepływ azotu (3.5) na ok. 60 pęcherzyków na minutę.
5.6. Wprowadzić 15 ml kwasu ortofosforowego (V) (3.2) z wkraplacza do kolby destylacyjnej A.
5.7. Ogrzewać szybko do wrzenia i następnie utrzymywać w stanie łagodnego wrzenia przez 30 minut.
5.8. Odłączyć odbiornik destylacji D. Popłukać rurkę łączącą i następnie miareczkować destylat roztworem wodorotlenku sodu (3.4) do zmiany zabarwienia wskaźnika na kolor zielony (3.6).
6. OBLICZENIA
Obliczenia zawartości siarczanu (IV) lub wodorosiarczanu (IV) w procentach masowych w próbce według wzoru:

gdzie:
M = molowe stężenie roztworu wodorotlenku sodu (3.4),
V = objętość roztworu wodorotlenku sodu (3.4) zużyta do miareczkowania (5.8), w mililitrach,
m = masa próbki (5.1), w gramach.
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości dwutlenku siarki 0,2% różnica między dwoma równoległymi oznaczeniami wykonanymi dla tej samej próbki nie powinna być większa niż 0,006%.
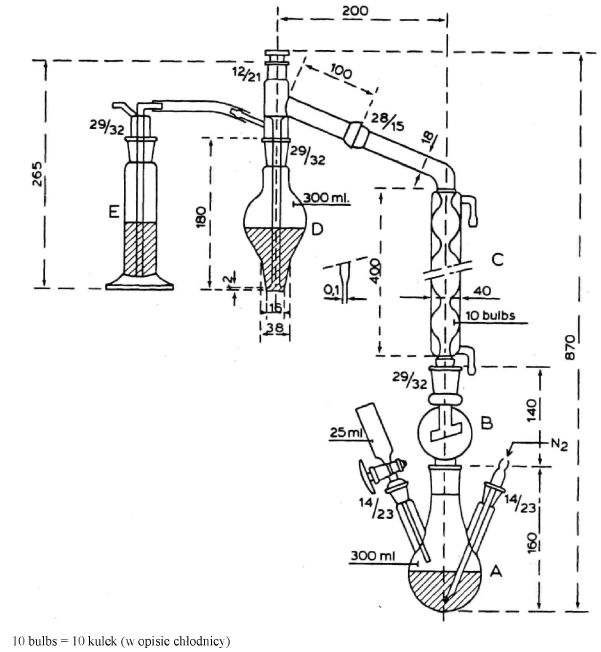
Rys. 12
Aparatura do destylacji dwutlenku siarki według Tannera
Wszystkie wymiary w mm
XXVIII. IDENTYFIKOWANIE I OZNACZANIE CHLORANÓW METALI ALKALICZNYCH
PRZEDMIOT I ZAKRES ZASTOSOWANIA
Metoda opisuje identyfikowanie i oznaczanie chloranów w pastach do zębów i innych produktach kosmetycznych.
A. IDENTYFIKOWANIE
1. ZASADA
Chlorany oddziela się od innych związków chlorowcowych przez chromatografię cienkowarstwową i identyfikuje przez utlenianie jodku potasu do jodu.
2. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
2.1. Roztwory odniesienia: wodne roztwory chloranu, bromianu i jodan potasu (0,2% m/v), świeżo przygotowane.
2.2. Rozpuszczalnik rozwijający: roztwór amoniaku (28% m/v) / aceton / butanol (60:130:30 obj.).
2.3. Jodek potasu, roztwór wodny (5% m/v).
2.4. Roztwór skrobi (od 1 do 5% m/v).
2.5. Kwas chlorowodorowy (1 M).
2.6. Przygotowane fabrycznie celulozowe płytki do chromatografii cienkowarstwowej (0,25 mm).
3. APARATURA
Zwykły sprzęt do chromatografii cienkowarstwowej.
4. PROCEDURA
4.1. Ekstrahować ok. 1 g próbki wodą, przesączyć i rozcieńczyć do ok. 25 ml.
4.2. Nanieść na płytkę (2.6) 2 µl roztworu (4.1) razem z 2 µl porcjami wszystkich trzech roztworów odniesienia (2.1).
4.3. Umieścić płytkę w komorze i rozwijać rozpuszczalnikiem (2.2) metodą wstępującej chromatografii do ok. trzech czwartych długości płytki (2.6).
4.4. Wyjąć płytkę z komory i odparować rozpuszczalnik. Uwaga: Może to trwać do dwóch godzin.
4.5. Spryskać płytkę roztworem jodku potasu (2.3) i pozostawić ją do wyschnięcia w ciągu ok. pięciu minut.
4.6. Spryskać płytkę roztworem skrobi (2.4) i pozostawić ją do wyschnięcia w ciągu ok. pięciu minut.
4.7. Spryskać płytkę kwasem chlorowodorowym (2.5).
5. OCENA
Jeżeli chloran jest obecny w próbce, to po upływie pół godziny pojawi się na płytce błękitna plama (może być także plama brązowa) o wartości Rf w przybliżeniu 0,7 do 0,8.
| Związek chlorowcowy | Rf |
| jodan bromian (V) chloran (V) | 0–0,2 0,5–0,6 0,7–0,8 |
Należy zauważyć, że bromiany i jodany ulegają reakcji natychmiast. Należy być ostrożnym, aby nie pomylić bromianów i chloranów.
B. OZNACZANIE
1. DEFINICJA
Zawartość chloranu w próbce oznaczona tą metodą jest wyrażona w procentach masowych chloranu.
2. ZASADA
Chloran ulega redukcji pyłem cynkowym w środowisku kwaśnym. Powstały chlorek mierzy się miareczkowaniem potencjometrycznym z użyciem roztworu azotanu srebra. Podobne oznaczenie przed redukcją pozwala określić możliwą obecność halogenków.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
3.1. Kwas octowy, 80% (m/m).
3.2. Pył cynkowy.
3.3. Mianowany roztwór azotanu srebra (0,1 M).
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Potencjometr wyposażony w elektrodę do wykrywania srebra.
5. PROCEDURA
5.1. Przygotowanie próbki
Zważyć dokładnie w probówce wirówki ilość „m” ok. 2 g próbki. Dodać ok. 15 ml kwasu octowego (3.1) i zamieszać starannie. Poczekać 30 minut i wirować w ciągu 15 minut przy 2000 obr./min. Zlać roztwór znad osadu do 50 ml kolby miarowej. Powtórzyć wirowanie dwukrotnie, dodając 15 ml kwasu octowego (3.1) do pozostałości. Zebrać roztwory zawierające chloran w tej samej kolbie miarowej. Uzupełnić do kreski kwasem octowym (3.1).
5.2. Redukcja chloranu
Pobrać 20 ml roztworu (5.1) i dodać 0,6 g pyłu cynkowego (3.2). Doprowadzić do wrzenia w kolbie wyposażonej w chłodnicę zwrotną. Po 30 minutach wrzenia schłodzić i przesączyć. Popłukać kolbę wodą. Przesączyć i połączyć z poprzednim przesączeniem.
5.3. Oznaczanie chlorku
Miareczkować 20 ml przesącza (5.2) azotanem srebra (3.3) z zastosowaniem potencjometru (4.2). W ten sam sposób miareczkować 20 ml roztworu (5.1) azotanem srebra (3.3).
Uwaga: Jeżeli produkt zawiera pochodne bromu lub jodu, które mogą uwalniać bromki lub jodki po redukcji, krzywa miareczkowania będzie miała kilka punktów przegięcia. W tym przypadku objętość roztworu azotanu srebra (3.3) użytego do miareczkowania odpowiadająca chlorkowi jest różnicą między ostatnim i przedostatnim punktem przegięcia.
6. OBLICZANIE
Zawartość chloranu w próbce (% m/m) oblicza się według wzoru:
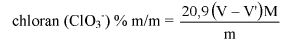
gdzie:
V = objętość w mililitrach roztworu azotanu srebra (3.3) zużyta do miareczkowania roztworu (5.2),
V' = objętość w mililitrach roztworu azotanu srebra (3.3) zużyta do miareczkowania 20 mililitrów roztworu (5.1),
M = molarność mianowanego roztworu azotanu srebra (3.3),
m = masa próbki, w gramach.
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości chloranu od 3 do 5% m/m różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,07% m/m.
XXIX. IDENTYFIKOWANIE I OZNACZANIE JODANU SODU
PRZEDMIOT I ZAKRES ZASTOSOWANIA
Metoda opisuje procedurę identyfikowania i oznaczania jodanu sodu (INCI: sodium iodate) w zmywalnych ze skóry i włosów produktach kosmetycznych, które go zawierają.
A. IDENTYFIKOWANIE
1. ZASADA
Jodan sodu oddziela się od innych związków chlorowcowych przez chromatografię cienkowarstwową i identyfikuje przez utlenianie jodku do utworzenia jodu.
2. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy.
2.1. Roztwory odniesienia: wodne roztwory chloranu, bromianu i jodanu potasu (0,01% m/v) świeżo przygotowane.
2.2. Roztwór rozwijający: roztwór amoniaku (28% m/v) / aceton / butanol (60:130:30 obj.).
2.3. Jodek potasu, wodny roztwór (5% m/v).
2.4. Roztwór skrobi (1 do 5% m/v).
2.5. Kwas chlorowodorowy.
3. APARATURA
3.1. Przygotowane fabrycznie celulozowe płytki do chromatografii cienkowarstwowej (0,25 mm).
3.2. Zwykły sprzęt do chromatografii cienkowarstwowej.
4. PROCEDURA
4.1. Ekstrahować ok. 1 g próbki wodą, przesączyć i rozcieńczyć do ok. 10 ml.
4.2. Nanieść 2 µl otrzymanego roztworu na linię bazową płytki (3.1) razem z 2 µl porcjami wszystkich trzech roztworów odniesienia (2.1).
4.3. Umieścić płytkę w komorze i rozwijać rozpuszczalnikiem (2.2) metodą wstępującej chromatografii do ok. trzech czwartych długości płytki.
4.4. Wyjąć płytkę z komory i pozostawić do odparowania w temperaturze otoczenia. Uwaga: Może to trwać do dwóch godzin.
4.5. Spryskać płytkę roztworem jodku potasu (2.3) i pozostawić do wyschnięcia na ok. pięć minut.
4.6. Spryskać płytkę roztworem skrobi (2.4) i pozostawić do wyschnięcia na ok. pięć minut.
4.7. Na końcu spryskać płytkę kwasem chlorowodorowym (2.5).
5. OCENA
Jeżeli w próbce jest obecny jodan, natychmiast pojawia się błękitna plama (zabarwienie może być brązowe lub stawać się brązowe z upływem czasu) o wartości Rf od 0,0 do 0,2.
Należy zauważyć, że bromiany dają natychmiastową barwną pianę o wartościach Rf od 0,5 do 0,6, a chlorany po ok. 30 minutach odpowiednio o wartościach Rf od 0,7 do 0,8.
B. OZNACZANIE
1. DEFINICJA
Zawartość jodanu sodu oznaczona tą metodą jest wyrażona jako procent masowy jodanu sodu.
2. ZASADA
Jodan sodu rozpuszcza się w wodzie i oznacza metodą wysokosprawnej chromatografii cieczowej, używając kolejno kolumny C18 z fazą odwróconą i kolumny z wymieniaczem anionowym.
3. ODCZYNNIKI
Wszystkie odczynniki powinny być czyste do analizy i szczególnie odpowiednie do wysokosprawnej chromatografii cieczowej (HPLC).
3.1. Kwas chlorowodorowy (4 M).
3.2. Siarczan sodu, wodny roztwór 5% m/v.
3.3. Jodan sodu, roztwór bazowy. Przygotować roztwór bazowy zawierający 50 mg jodanu sodu w 100 ml wody.
3.4. Diwodoroortofosforan potasu.
3.5. 2-hydrat wodoroortofosforanu (V) disodu (NaHPO4 2H2O).
3.6. Faza ruchoma HPLC: rozpuścić 3,88 g diwodoroortofosforanu (V) potasu (3.4) i 1,19 g 2-hydratu wodoroortofosforanu (V) disodu (3.5) w 1 litrze wody.
Otrzymany roztwór ma pH 6,2.
3.7. Uniwersalny papierek wskaźnikowy pH 1–11.
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Bibuła filtracyjna, krążki o średnicy 110 mm, Schleicher and Schüll nr 575 lub równoważna.
4.3. Chromatograf do wysokosprawnej chromatografii cieczowej z detektorem o zmiennej długości fali.
4.4. Kolumny: długość 120 mm, średnica wewnętrzna 4,6 mm; dwie kolumny połączone kolejno: pierwsza kolumna – NecleosilR 5 C18 lub równoważna; druga kolumna – Vydac™-301-SB lub równoważna.
5. PROCEDURA
5.1. Przygotowanie próbki
5.1.1. Próbki ciekłe (szampony).
W 10 ml szklanej probówce wyskalowanej i zamykanej korkiem lub kolbie miarowej zważyć dokładnie odważkę analityczną ok. 1,0 g próbki.
Uzupełnić wodą do kreski i wymieszać.
Przesączyć roztwór, jeżeli potrzeba.
Oznaczyć jodan w roztworze metodą HPLC zgodnie z opisem w pkt 5.2.
5.1.2. Próbki stałe (mydła)
Rozdrobnić starannie część próbki i zważyć odważkę analityczną ok. 1,0 g w 100 ml szklanym cylindrze miarowym z korkiem. Uzupełnić wodą do 50 ml i wytrząsać energicznie w ciągu jednej minuty. Odwirować i przesączyć przez bibułę filtracyjną (4.2) i pozostawić mieszaninę do odstania co najmniej do następnego dnia. Wytrząsnąć podobny do żelu roztwór i przesączyć go przez bibułę filtracyjną (4.2). Oznaczyć jodan w przesączu metodą HPLC zgodnie z opisem w pkt 5.2.
5.2. Chromatografia
Przepływ: 1 ml/min.
Długość fali detektora (4.3): 210 nm.
Wstrzykiwana objętość próbki: 10 µl.
Sposób pomiaru: obliczenie wielkości powierzchni piku.
5.3. Kalibrowanie
Do 50 ml kolb miarowych wprowadzić pipetą kolejno po 1,0; 2,0; 5,0; 10,0 i 20,0 ml bazowego roztworu jodanu sodu (3.3).
Uzupełnić do kreski i wymieszać. Otrzymane w ten sposób roztwory zawierają odpowiednio 0,01; 0,02; 0,05; 0,10 i 0,20 mg jodanu sodu na ml. Wprowadzić strzykawką kolejno 10 µl porcje wszystkich standardowych roztworów jodanu do chromatografu (4.3) i otrzymać ich chromatogramy. Oznaczyć powierzchnie pików dla jodanu i wykreślić krzywą przedstawiającą stosunek powierzchni piku do stężenia jodanu sodu.
6. OBLICZANIE
Obliczyć zawartość jodanu sodu w procentach masowych (% m/m) według wzoru:

gdzie:
m = masa w gramach odważki analitycznej (5.1),
V = całkowita objętość roztworu próbki, w mililitrach, otrzymanego jak opisano w pkt 5.1,
c = stężenie, w miligramach na mililitr jodanu sodu, otrzymane z krzywej kalibracyjnej (5.3).
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości jodanu sodu 0,1% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonywanych dla tej samej próbki nie może przekraczać 0,002%.
8. POTWIERDZENIE
8.1. Zasada
W kwaśnym roztworze produktu kosmetycznego jodan sodu (IO3 -) jest zredukowany do jodku (I-) przez siarczan i powstający w rezultacie roztwór jest badany metodą HPLC. Jeżeli szczyt, przy czasie retencji korespondującym z czasem retencji jodanu, zanika po zastosowaniu siarczanu, pierwotny szczyt można uznać za efekt działania jodanu.
8.2. Procedura
Wprowadzić pipetą do kolby stożkowej 5 ml porcję roztworu próbki otrzymanego, jak opisano w pkt 5.1.
Doprowadzić pH roztworu do wartości 3 lub niższej poprzez dodanie kwasu chlorowodorowego (3.1). Sprawdzić wartość pH uniwersalnym papierkiem wskaźnikowym (3.7).
Dodać trzy krople roztworu siarczanu sodu (3.2) i wymieszać. Wprowadzić strzykawką 10 µl porcję roztworu do chromatografu cieczowego (4.3). Porównywać ten ostatni chromatogram z chromatogramem otrzymanym, jak opisano w pkt 5 dla tej samej próbki.
XXX. IDENTYFIKOWANIE I OZNACZANIE AZOTANU (V) SREBRA W PRODUKTACH KOSMETYCZNYCH
A. IDENTYFIKOWANIE
1. CEL I ZAKRES STOSOWANIA
Celem metody jest identyfikowanie azotanu (V) srebra (INCI: silver nitrate) jako srebra w produktach kosmetycznych zawierających wodę.
2. ZASADA
Srebro identyfikuje się dzięki tworzeniu białego, charakterystycznego osadu z jonami chloru.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Kwas solny, roztwór 2 M.
3.2. Roztwór amoniaku: rozcieńczyć stężony roztwór wodorotlenku amonu (d20 = 0,88 g/ml) równą ilością wody i wymieszać.
3.3. Kwas azotowy (V), roztwór 2 M.
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Wirówka.
5. PROCEDURA
5.1. Do ok. 1 g próbki w probówce wirówki dodawać kroplami 2 M roztwór kwasu solnego (3.1) aż do całkowitego wytrącenia osadu; wymieszać i odwirować.
5.2. Odrzucić ciecz znajdującą się na powierzchni i przemyć jednokrotnie osad pięcioma kroplami zimnej wody. Wylać przemywki.
5.3. Dodać wodę w ilości równej masie osadu w probówce wirówki. Ogrzewać do wrzenia i wymieszać.
5.4. Odwirować gorącą próbkę, odrzucić ciecz znajdującą się na wierzchu.
5.5. Do osadu dodać kilka kropli roztworu amoniaku (3.2); wymieszać i odwirować.
5.6. Do jednej kropli cieczy znajdującej się na szklanej płytce dodać kilka kropli 2 M roztworu kwasu azotowego (V) (3.3).
5.7. Biały osad wskazuje na obecność srebra.
B. OZNACZANIE
1. CEL I ZAKRES
Metoda jest odpowiednia dla oznaczania azotanu (V) srebra jako srebra w produktach kosmetycznych przeznaczonych do barwienia brwi i rzęs.
2. ZASADA
Srebro oznacza się w produkcie metodą absorpcyjnej spektrometrii atomowej.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Kwas azotowy (V), roztwór 0,02 M.
3.2. Roztwory mianowane srebra.
3.2.1. Bazowy roztwór mianowany srebra zawierający 1000 µg/ml w 0,5 M roztworze kwasu azotowego (V) (SpectrosoL lub równorzędny).
3.2.2. Roztwór mianowany srebra zawierający 100 µg/ml: przenieść pipetą 10 ml bazowego roztworu mianowanego srebra (3.2.1) do 100 ml kolby miarowej. Uzupełnić objętość 0,02 M roztworem kwasu azotowego (V) (3.1) i wymieszać. Mianowany roztwór powinien być świeży i przechowywany w butelce z ciemnego szkła.
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny
4.2. Spektrofotometr absorpcji atomowej wyposażony w lampę z katodą wnękową do oznaczania srebra.
5. PROCEDURA
5.1. Przygotowanie próbki
Odważyć dokładnie ok. 0,1 g (m gramów) jednolitej (homogenicznej) próbki produktu. Przenieść ilościowo odważkę do jednolitrowej kolby miarowej. Uzupełnić objętość 0,02 M roztworem kwasu azotowego (V) (3.1) i wymieszać.
5.2. Warunki analizy metodą absorpcyjnej spektometrii atomowej:
płomień: powietrze – acetylen
długość fali: 338,3 nm
korekta tła: stosuje się
własności płomienia: ubogi, dla uzyskania maksymalnej absorbancji konieczna będzie optymalizacja wysokości palnika i warunków spalania.
5.3. Kalibrowanie
5.3.1. Przenieść pipetą po 1,0; 2,0; 3,0; 4,0 i 5,0 ml roztworu mianowanego srebra (3.2.2) do serii 100 ml kolb miarowych, uzupełnić objętość w każdej kolbie do kreski 0,02 M roztworem kwasu azotowego (3.1) i wymieszać. Roztwory zawierają odpowiednio 1,0; 2,0; 3,0; 4,0 i 5,0 mg srebra w milimetrze.
5.3.2. Zmierzyć absorbancję 0,02 M roztworu kwasu azotowego (V) (3.1) i stosować uzyskaną wartość jako odpowiadającą zerowemu stężeniu srebra dla krzywej odwzorowania. Zmierzyć absorbancję każdego roztworu mianowanego srebra do odwzorowania (5.3.1). Wykreślić krzywą odwzorowania odpowiadającą stosunkom wartości absorbancji do stężenia srebra.
5.4. Oznaczanie
Zmierzyć absorbancję roztworu próbki (5.1). Z krzywej odwzorowania odczytać stężenie srebra odpowiadające wartości absorbancji otrzymanej dla roztworu próbki.
6. OBLICZENIE
Obliczenie zawartości azotanu (V) srebra w próbce, w procentach masowych (% mm), według wzoru:
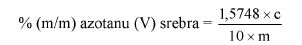
gdzie:
m = masa w gramach próbki pobranej do analizy (5.1),
c = stężenie srebra w roztworze próbki (5.1), w mikrogramach na mililitr, otrzymane w krzywej odwzorowania.
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości azotanu (V) srebra wynoszącej 4% (m/m) różnica między wynikami dwóch oznaczeń wykonanych równolegle dla tej samej próbki nie powinna przekraczać 0,05% (m/m).
XXXI. IDENTYFIKOWANIE I OZNACZANIE DISIARCZKU SELENU W SZAMPONACH PRZECIWŁUPIEŻOWYCH
A. IDENTYFIKOWANIE
1. CEL I ZAKRES STOSOWANIA
Celem metody jest identyfikowanie disiarczku selenu jako selenu w szamponach przeciwłupieżowych.
2. ZASADA
Selen identyfikuje się przez charakterystyczny kolor od żółtego do pomarańczowego powstający w reakcji z mocznikiem i jodkiem potasu.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Kwas azotowy (V) stężony, d20= 1,42 g/ml.
3.2. Mocznik.
3.3. Jodek potasu, roztwór 10% (m/v); rozpuścić 10 g jodku potasu w 100 ml wody.
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Rurka do wytrawiania, 100 ml objętości.
4.3. Ogrzewany blok do wytrawiania.
4.4. Bibuła filtracyjna (Whatman nr 42 lub równoważna) lub filtr membranowy o średnicy porów 0,45 µm.
5. PROCEDURA
5.1. Do ok. 1 g szamponu w rurce do trawienia (4.2) dodać 2,5 ml stężonego kwasu azotowego (V) (3.1) i wytrawić w temperaturze 150°C w ciągu 30 minut w ogrzewanym bloku do wytrawienia (4.3).
5.2. Rozcieńczyć wytrawioną próbkę wodą do 25 ml i przesączyć przez bibułę filtracyjną lub filtr membranowy o średnicy porów 0,45 mm (4.4).
5.3. Do 2,5 ml przesączu dodać 5 ml wody, 2,5 g mocznika (3.2) i ogrzewać do wrzenia. Schłodzić i dodać 1 ml roztworu jodku potasu (3.3).
5.4. Zabarwienie od żółtego do pomarańczowego, które ciemnieje podczas przechowywania, wskazuje na obecność selenu.
B. OZNACZANIE
1. CEL I ZAKRES
Celem metody jest oznaczanie disiarczku selenu jako selenu w szamponach przeciwłupieżowych zawierających do 4,5% (m/m) disiarczku selenu.
2. ZASADA
Próbka jest wytrawiona kwasem azotowym (V). W powstałym roztworze selen jest oznaczony za pomocą absorpcyjnej spektometrii atomowej.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Kwas azotowy (V) stężony, d20 = 1,42 g/ml.
3.2. Kwas azotowy (V), roztwór 5% (obj.): dodać ze stałym mieszaniem 50 ml stężonego kwasu azotowego (V) (3.1) do 500 ml wody w zlewce. Przenieść ten roztwór do jednolitrowej kolby miarowej i uzupełnić objętość wodą do kreski.
3.3. Bazowy mianowany roztwór selenu zawierający 1000 µg/ml w 0,5 M kwasie azotowym (V) (SpectrosoL lub równoważny).
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Rurka do wytrawiania, 100 ml objętości.
4.3. Ogrzewany blok do wytrawiania.
4.4. Bibuła filtracyjna (Whatman nr 42 lub równoważna) lub filtr membranowy o średnicy porów 0,45 mm.
4.5. Spektrofotometr absorpcji atomowej wyposażony w lampę z katodą wnękową do oznaczania selenu.
5. PROCEDURA
5.1. Przygotowanie próbki
5.1.1. Zważyć dokładnie ok. 0,2 g (m gramów) jednolitej (homogenicznej) próbki szamponu w rurce do wytrawiania (4.2).
5.1.2. Dodać 5 ml stężonego kwasu azotowego (V) (3.1) i wytrawiać w temperaturze 150°C w ciągu jednej godziny w ogrzewanym bloku do wytrawiania (4.3).
5.1.3. Pozostawić roztwór do schłodzenia i rozcieńczyć wodą do 100 ml. Przesączyć przez bibułę filtracyjną lub filtr membranowy o średnicy porów 0,45 mm (4.4) i zachować przesącz do wykonania oznaczenia.
5.2. Warunki analizy metodą absorpcyjnej spektrometrii atomowej: płomień:
powietrze – acetylen
długość fali: 196,0 nm
korekcja tła: stosuje się
własności płomienia: ubogi, dla uzyskania maksymalnej absorbancji konieczna będzie optymalizacja wysokości palnika i warunków spalania.
5.3. Kalibrowanie
5.3.1. Przenieść pipetą po 1,0, 2,0, 3,0, 4,0 i 5,0 ml bazowego roztworu mianowanego selenu (3.3) do serii 100 ml kolb miarowych. Uzupełnić objętość we wszystkich kolbach do kreski 5% (obj.) roztworem kwasu azotowego (V) (3.2) i wymieszać. Roztwory te zawierają odpowiednio po 10, 20, 30, 40 i 50 µg selenu w mililitrze.
5.3.2. Zmierzyć absorbancję 5% (obj.) roztworu kwasu azotowego (V) (3.2) i stosować uzyskaną wartość jako odpowiadającą zerowemu stężeniu selenu dla krzywej odwzorowania. Zmierzyć absorbancję wszystkich mianowanych roztworów selenu do kalibrowania (5.3.1). Wykreślić krzywą odwzorowania odpowiadającą stosunkom wartości absorbancji do stężenia selenu.
5.4. Oznaczenie
Zmierzyć absorbancję roztworu próbki (5.1.3). Z krzywej odwzorowania odczytać stężenie selenu odpowiadające wartości absorbancji otrzymanej dla roztworu próbki.
6. OBLICZENIE
Obliczyć zawartość disiarczku selenu w próbce, w procentach masowych (% m/m), według wzoru:

gdzie:
m = masa w gramach próbki pobranej do analizy (5.1),
c = stężenie selenu w roztworze próbki (5.1.3), w mikrogramach na mililitr, otrzymane z krzywej odwzorowania.
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości selenu wynoszącej 1% (m/m) różnica między wynikami dwóch oznaczeń wykonanych równolegle dla tej samej próbki nie powinna przekraczać 0,05% (m/m).
XXXII. OZNACZANIE ROZPUSZCZALNYCH POSTACI BARU I STRONTU W PIGMENTACH W FORMIE SOLI I LAKÓW
A. OZNACZANIE ROZPUSZCZALNEGO BARU
1. CEL I ZAKRES STOSOWANIA
Metoda podaje procedurę ekstrakcji i oznaczania rozpuszczalnego baru z pigmentów w formie soli i laków.
2. ZASADA
Pigment ekstrahuje się 0,07 M roztworem kwasu solnego w określonych warunkach i ilość baru w ekstrakcie oznacza się metodą absorpcyjnej spektrometrii atomowej.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Alkohol etylowy absolutny.
3.2. Kwas solny, roztwór 0,07 M.
3.3. Kwas solny, roztwór 0,5 M.
3.4. Chlorek potasu, roztwór 8% (m/m); rozpuścić 16 g chlorku potasu w 200 ml 0,07 M roztworu kwasu solnego (3.2).
3.5. Mianowany roztwór baru.
3.5.1. Bazowy mianowany roztwór baru zawierający 1000 µg/ml w 0,5 M roztworze kwasu azotowego (V) (SpectrosoL lub równoważny).
3.5.2. Mianowany roztwór baru zawierający 200 µg/ml: przenieść pipetą 20,0 ml bazowego roztworu mianowanego baru (3.5.1) do 100 ml kolby miarowej. Uzupełnić objętość do kreski 0,07 M roztworem kwasu solnego (3.2) i wymieszać.
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Pehametr o dokładności +/– 0,02 jednostek.
4.3. Przegubowa wytrząsarka do kolb.
4.4. Filtr membranowy o średnicy porów 0,45 mm.
4.5. Spektrofotometr absorpcji atomowej wyposażony w lampę z katodą wnękową do oznaczania baru.
5. PROCEDURA
5.1. Przygotowanie próbki
5.1.1. Odważyć dokładnie ok. 0,5 g (m gramów) pigmentu w kolbie stożkowej.
Dla zapewnienia wystarczającej objętości do energicznego mieszania nie należy używać kolb o objętości mniejszej niż 150 ml.
5.1.2. Dodać pipetą 1,0 ml alkoholu etylowego (3.1) i obracać kolbę, aby zapewnić dokładne zwilżenie pigmentu. Dodać z biurety oznaczoną ilość 0,07 M roztworu kwasu solnego (3.2) wymaganą dla otrzymania stosunku „objętości kwasu” do „masy pigmentu” wynoszącej dokładnie 50 mililitrów na gram. Oznaczyć całkowitą objętość ekstraktu łącznie z alkoholem etylowym jako „V” w ml. Wymieszać zawartość kolby ruchem wirowym w ciągu pięciu sekund dla zapewnienia dokładnego wymieszania zawartości.
5.1.3. Za pomocą pehametru (4.2) zmierzyć pH otrzymanej suspensji i jeżeli wynosi ono powyżej 1,5, dodać kroplami 0,5 M roztwór kwasu solnego (3.3) aż do uzyskania pH 1,4–1,5.
5.1.4. Zamknąć kolbę korkiem i natychmiast wytrząsnąć w ciągu 60 minut w wytrząsarce przegubowej do kolb (4.3). Wytrząsarka musi działać z wystarczająco dużą szybkością, tak aby wytworzyła się piana. Przesączyć przez filtr membranowy o średnicy porów 0,45 mm (4.4) i zebrać przesącz. Nie należy wirować ekstraktu przed przesączeniem. Przenieść pipetą 5,0 ml przesączu do 50 ml kolby miarowej; uzupełnić objętość do kreski 0,07 M kwasu solnego (3.2) i wymieszać. Tego roztworu używa się do oznaczania strontu (dział B).
5.1.5. Do 100 ml kolby miarowej przenieść pipetą 5,0 ml roztworu chlorku potasu (3.4) i pewną objętość (WBa ml) rozcieńczonego przesączu (5.1.4) dla otrzymania spodziewanego stężenia pomiędzy 3 i 10 mg baru na mililitr (objętość 10 ml powinna być zadawalającym punktem wyjściowym). Uzupełnić objętość w kolbie do kreski 0,07 M roztworem kwasu solnego (3.2) i wymieszać.
5.1.6. Tego samego dnia oznaczyć stężenie baru w roztworze (5.1.5) metodą absorpcyjnej spektrometrii atomowej.
5.2. Warunki analizy metodą absorpcyjnej spektrometrii atomowej:
płomień: podtlenek azotu/acetylen
długość fali: 553,5 nm
korekcja tła: nie stosuje się
własności płomienia: ubogi, dla uzyskania maksymalnej absorbancji konieczna będzie optymalizacja wysokości palnika i warunków spalania.
5.3. Kalibrowanie
5.3.1. Przenieść pipetą do 1,0, 2,0, 3,0, 4,0 i 5,0 ml roztworu mianowanego baru (3.5.2) do serii 100 ml kolb miarowych. Do każdej kolby dodać pipetą 5,0 ml roztworu chlorku potasu (3.4); uzupełnić objętość do kreski 0,07 M roztworem kwasu solnego (3.2) i wymieszać. Roztwory te zawierają odpowiednio 2,0, 4,0, 6,0, 8,0 i 10 mg baru na mililitr. Ślepą próbę przygotować podobnie, bez dodawania roztworu mianowanego baru.
5.3.2. Zmierzyć absorbancję ślepej próby (5.3.1) i stosować uzyskaną wartość jako odpowiadającą zerowemu stężeniu baru dla krzywej odwzorowania. Zmierzyć absorbancję wszystkich mianowanych roztworów baru do kalibrowania (5.3.1) Wykreślić krzywą odwzorowania odpowiadającą stosunkom wartości absorbancji do stężenia baru.
5.4. Oznaczanie
Zmierzyć absorbancję roztworu próbki (5.1.5). Z krzywej odwzorowania odczytać stężenie baru odpowiadające wartości absorbancji otrzymanej dla roztworu próbki.
6. OBLICZENIE
Zawartość rozpuszczalnego baru (% m/m) w pigmencie oblicza się według wzoru:
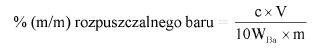
gdzie:
m = masa w gramach próbki pobranej do analizy (5.1.1),
c = stężenie baru w roztworze próbki (5.1.5), w mikrogramach na mililitr, otrzymane z krzywej odwzorowania,
V = całkowita objętość ekstraktu w mililitrach (5.1.2),
WBa = objętość ekstraktu w mililitrach (5.1.5).
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Najlepsza dostępna ocena powtarzalności dla tej metody wynosi 0,3% dla zawartości rozpuszczalnego baru wynoszącej 2% (m/m).
8. UWAGI
8.1. W pewnych warunkach absorbancja baru może wzrosnąć w obecności wapnia. Temu wzrostowi można przeciwdziałać, dodając jon magnezu w stężeniu 5g na litr.
8.2. Zastosowanie metody plazmy sprzężonej indukcyjnie – emisyjnej spektometrii atomowej jest dozwolone jako metody alternatywnej w stosunku do płomieniowej absorpcyjnej spektrometrii atomowej.
B. OZNACZANIE ROZPUSZCZALNEGO STRONTU
1. CEL I ZAKRES STOSOWANIA
Metoda podaje procedurę ekstrakcji i oznaczania rozpuszczalnego strontu z pigmentów w formie soli lub laków.
2. ZASADA
Pigment ekstrahuje się 0,07 M roztworem kwasu solnego w określonych warunkach, a ilość strontu w ekstrahentach oznacza się metodą absorpcyjnej spektrometrii atomowej.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Alkohol etylowy absolutny.
3.2. Kwas solny, 0,07 M roztwór.
3.3. Chlorek potasu, roztwór 8% (m/v); rozpuścić 16 g chlorku potasu w 200 ml 0,07 M roztworu kwasu solnego (3.2).
3.4. Mianowane roztwory strontu.
3.4.1. Bazowy mianowany roztwór strontu zawierający 1000 µg/ml w 0,5 M roztworze kwasu azotowego (V) (SpectrosoL lub równoważny).
3.4.2. Mianowany roztwór strontu zawierający 100 µg/ml: przenieść pipetą 10,0 ml bazowego roztworu mianowanego strontu (3.4.1) do 100 ml kolby miarowej. Uzupełnić objętość do kreski 0,07 M roztworem kwasu solnego (3.2) i wymieszać.
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Filtr membranowy o średnicy porów 0,45 mm.
4.3. Spektrofotometr absorpcji atomowej wyposażony w lampę z katodą wnękową do oznaczania strontu.
5. PROCEDURA
5.1. Przygotowanie próbki
Roztworu przygotowanego według opisu w A.5.1.4 używa się do oznaczania zawartości rozpuszczalnego strontu.
5.1.1. Do 100 ml kolby miarowej przenieść pipetą 5,0 ml roztworu chlorku potasu (3.3) i pewną objętość (Wsr ml) rozcieńczonego przesączu (A.5.1.4) do otrzymania oczekiwanego stężenia między 2 i 5 µg strontu na mililitr (objętość 25 ml powinna być zadawalającym punktem wyjściowym). Uzupełnić objętość do kreski 0,07 M roztworem kwasu solnego (3.2) i wymieszać.
5.1.2. Oznaczyć stężenie strontu w roztworze (5.1.1) metodą absorpcyjnej spektrometrii atomowej tego samego dnia.
5.2. Warunki analizy metodą absorpcyjnej spektometrii atomowej:
płomień: podtlenek azotu / acetylen
długość fali: 460,7 nm
korekcja tła: nie stosuje się
własności płomienia: ubogi, dla uzyskania maksymalnej absorbancji konieczna będzie optymalizacja wysokości palnika i warunków spalania.
5.3. Kalibrowanie
5.3.1. Przenieść pipetą po 1,0, 2,0, 3,0, 4,0 i 5,0 ml roztworu mianowanego strontu (3.4.2) do serii 100 ml kolb miarowych. Do wszystkich kolb dodać pipetą po 5,0 ml roztworu chlorku potasu (3.3); uzupełnić objętość do kreski 0,07 M roztworem kwasu solnego (3.2) i wymieszać. Roztwory te zawierają odpowiednio 1,0; 2,0; 4,0 i 5,0 µg strontu w mililitrze. Podobnie przygotować ślepą próbę bez dodawania roztworu mianowanego strontu.
5.3.2. Zmierzyć absorbancję roztworu ślepej próby (5.3.1) i stosować uzyskaną wartość jako odpowiadającą zerowemu stężeniu stromi dla krzywej odwzorowania odpowiadającej stosunkom wartości piku absorbancji do stężenia strontu.
5.4. Oznaczanie
Zmierzyć absorbancję roztworu próbki (5.1.1). Z krzywej odwzorowania odczytać stężenie strontu odpowiadające wartości absorbancji otrzymanej dla roztworu próbki.
6. OBLICZENIE
Zawartość rozpuszczalnego strontu (% m/m) w pigmencie oblicza się według wzoru:
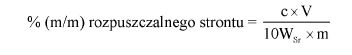
gdzie:
m = masa próbki pobranej do analizy (A.5.1.1) w gramach,
c = stężenie strontu w roztworze próbki (5.1.1) otrzymane z krzywej odwzorowania, w mikrogramach na mililitr,
V = objętość ekstraktu w mililitrach (A.5.1.2),
W = objętość ekstraktu w mililitrach (5.1.1).
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Najlepsza dostępna ocena powtarzalności dla tej metody wynosi 0,09% dla zawartości rozpuszczalnego strontu wynoszącej 0,6% (m/m).
8. UWAGA
Zastosowanie metody plazmy sprzężonej indukcyjnie – emisyjnej spektometrii atomowej jest dozwolone jako metody alternatywnej w stosunku do płomieniowej absorpcyjnej spektrometrii atomowej.
XXXIII. IDENTYFIKOWANIE I OZNACZANIE ALKOHOLU BENZYLOWEGO W PRODUKTACH KOSMETYCZNYCH
A. IDENTYFIKOWANIE
1. CEL I ZAKRES STOSOWANIA
Celem metody jest identyfikowanie alkoholu benzylowego w produktach kosmetycznych.
2. ZASADA
Alkohol benzylowy identyfikuje się metodą chromatografii cienkowarstwowej na płytkach z żelem krzemionkowym.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Alkohol benzylowy.
3.2. Chloroform.
3.3. Alkohol etylowy absolutny.
3.4. n-pentan.
3.5. Rozpuszczalnik rozwijający: eter dietylowy.
3.6. Mianowany roztwór alkoholu benzylowego: odważyć 0,1 g alkoholu benzylowego (3.1) w 100 ml kolbie miarowej, uzupełnić objętość do kreski alkoholem etylowym (3.3) i wymieszać.
3.7. Płytki do chromatografii cienkowarstwowej, szklane, 100 x 200 mm lub 200 x 200 mm, pokryte warstwą żelu krzemionkowego o grubości 0,25 mm.
3.8. Środek wywołujący: kwas dodekamolibdenianofosforowy 10% (m/v) roztwór w alkoholu etylowym (3.3).
4. APARATURA
4.1. Zwykła aparatura do chromatografii cienkowarstwowej.
4.2. Zbiornik chromatograficzny, komora z dwoma zagłębieniami, o ogólnych przybliżonych wymiarach 80 mm x 230 mm x 240 mm.
4.3. Bibuła chromatograficzna: Whatman lub równoważna.
4.4. Lampa o promieniowaniu w zakresie nadfioletu, długość fali 254 nm.
5. PROCEDURA
5.1. Przygotowanie próbki
W 10 ml kolbie miarowej zważyć 1,0 g analizowanego produktu. Dodać 3 ml chloroformu (3.2) i wytrząsać energicznie aż do zdyspergowania produktu. Uzupełnić objętość alkoholem etylowym (3.3) do kreski i wytrząsnąć energicznie do powstawania klarownego lub prawie klarownego roztworu.
5.2. Chromatografia cienkowarstwowa.
5.2.1. Wysycać komorę chromatograficzną (4.2) n-pentanem następująco: wyłożyć ścianki komory chromatograficznej przylegające do ściany tylnego zagłębienia bibułą chromatograficzną (4.3), upewniając się, że dolny brzeg bibuły znajduje się w zagłębieniu. Przenieść 25 ml n-pentanu (3.4) do tylnego zagłębienia, nalewając ten rozpuszczalnik ponad widoczną powierzchnię bibuły chromatograficznej wykładającej ściany. Natychmiast zamknąć pokrywę i pozostawić komorę na 15 minut.
5.2.2. Nanieść 10 ml roztworu próbki (5.1) i 10 ml roztworu mianowanego alkoholu benzylowego (3.6) w odpowiednich punktach linii startowej płytki do chromatografii cienkowarstwowej (3.7). Pozostawić do wyschnięcia.
5.2.3. Do przedniego zagłębienia komory wprowadzić pipetą 10 ml eteru dietylowego (3.5) i natychmiast umieścić płytkę (5.2.2) w tym samym zagłębieniu. Szybko zamknąć pokrywę komory i rozwijać chromatogram na płytce na odległość 150 mm. Usunąć płytkę z komory chromatograficznej i pozostawić do wyschnięcia w temperaturze pokojowej.
5.2.4. Obserwować płytkę (5.2.3) w świetle nadfioletowym i zaznaczyć położenie fioletowych plam. Spryskać płytkę środkiem wywołującym (3.8) i następnie ogrzewać płytkę w temperaturze 120°C w ciągu ok. 15 minut. Alkohol benzylowy występuje jako ciemnoniebieska plama.
5.2.5. Obliczyć wartość Rf otrzymaną dla roztworu mianowanego alkoholu benzylowego. Ciemnoniebieska plama o tym samym czasie retencji Rf otrzymana z roztworu próbki wskazuje na obecność alkoholu benzylowego. Granica wykrywalności: 0,1 µg alkoholu benzylowego.
B. OZNACZANIE
1. CEL I ZAKRES STOSOWANIA
Celem metody jest oznaczanie alkoholu benzylowego w produktach kosmetycznych.
2. DEFINICJA
Ilość alkoholu benzylowego oznaczana tą metodą jest wyrażona w procentach masowych (% m/m).
3. ZASADA
Próbkę ekstrahuje się metanolem i ilość alkoholu benzylowego w ekstrakcie oznacza się metodą wysokosprawnej chromatografii cieczowej (HPLC).
4. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
4.1. Metanol.
4.2. 4-etoksyfenol.
4.3. Alkohol benzylowy.
4.4. Faza ruchoma: metanol (4.1), woda (45:55 obj.).
4.5. Bazowy roztwór alkoholu benzylowego: w 100 ml kolbie miarowej dokładnie odważyć ok. 0,1 g alkoholu benzylowego (4.3). Uzupełnić objętość metanolem (4.1) do kreski i wymieszać.
4.6. Bazowy wewnętrzny roztwór mianowany: w 100 ml kolbie miarowej zważyć dokładnie ok. 0,1 g 4-etoksyfenolu (4.2). Uzupełnić objętość metanolem (4.1) do kreski i wymieszać.
4.7. Roztwory mianowane: do serii 25 ml kolb miarowych wprowadzić pipetą podane w poniższej tabeli ilości bazowego roztworu alkoholu benzylowego (4.5) i bazowego wewnętrznego roztworu mianowanego (4.6). Uzupełnić objętość do kreski metanolem (4.1) i wymieszać.
| Roztwór mianowany | Stężenie alkoholu benzylowego | Stężenie 4-etoksyfenolu | ||
| Dodana ilość, w ml (4.5) | µg/ml* | Dodana ilość, w ml (4.6) | µg/ml* | |
| I | 0,5 | 20 | 2,0 | 80 |
| II | 1,0 | 40 | 2,0 | 80 |
| III | 2,0 | 80 | 2,0 | 80 |
| IV | 3,0 | 120 | 2,0 | 80 |
| V | 5,0 | 200 | 2,0 | 80 |
| * Wartości te podano jako wskazówkę i odpowiadają one stężeniom roztworów mianowanych przygotowanych z użyciem roztworów alkoholu benzylowego (4.5) i 4-etokstyfenolu (4.6), które zawierają odpowiednio ściśle 0,1% (m/v) alkoholu benzylowego i ściśle 0,1% (m/v) 4-etoksyfenolu. | ||||
5. APARATURA
5.1. Zwykły sprzęt laboratoryjny.
5.2. Zestaw do wysokosprawnej chromatografii cieczowej z detektorem w zakresie długości fal ultrafioletowych o zmiennej długości fali i objętością pętli przy wstrzykiwaniu 10 µl próbki.
5.3. Chromatograficzna kolumna analityczna: 250 mm x 4,6 mm ze stali kwasoodpornej wypełniona 5 µm Sphedsorb ODS lub równoważnym wypełnieniem.
5.4. Łaźnia wodna.
5.5. Łaźnia ultradźwiękowa.
5.6. Wirówka.
5.7. Probówki do wirówki o objętości 15 ml.
6. PROCEDURA
6.1. Przygotowanie próbki
6.1.1. W probówce wirówki (5.7) odważyć dokładnie ok. 0,1 g (m gramów) próbki i dodać 5 ml metanolu (4.1).
6.1.2. Ogrzewać w łaźni wodnej utrzymywanej w temperaturze 50°C w ciągu 10 minut, następnie umieścić probówkę w łaźni ultradźwiękowej (5.5) aż do czasu, kiedy próbka będzie dokładnie zdyspergowana.
6.1.3. Schłodzić, następnie wirować próbkę przy 3500 obr./min w ciągu pięciu minut.
6.1.4. Przenieść ciecz znajdującą się na wierzchu do 25 ml kolby miarowej.
6.1.5. Ponownie ekstrahować próbkę dalszymi 5 ml metanolu (4.1). Połączyć ekstrakty w 25 ml kolbie miarowej.
6.1.6. Przenieść pipetą do 25 ml kolby miarowej 2,0 ml bazowego wewnętrznego roztworu mianowanego (4.6). Uzupełnić objętość metanolem (4.1) do kreski i wymieszać. Ten roztwór jest używany na etapie oznaczania analizy (6.4).
6.2. Chromatografia
6.2.1. Przygotować zestaw do wysokosprawnej chromatografii cieczowej (5.2) w zwykły sposób. Nastawić szybkość przepływu fazy ruchomej (4.4) na 2,0 ml na minutę.
6.2.2. Nastawić długość fali w detektorze w zakresie fal ultrafioletowych (5.2) na 210 nm.
6.3. Kalibrowanie
6.3.1. Wprowadzić strzykawką kolejno po 10 ml wszystkich mianowanych roztworów alkoholu benzylowego (4.7) i zmierzyć powierzchnie pików alkoholu benzylowego i 4-etoksyfenolu.
6.3.2. Dla każdego roztworu mianowanego alkoholu benzylowego (4.7) obliczyć stosunek powierzchni pików alkoholu benzylowego do powierzchni pików 4-etoksyfenolu. Wykreślić krzywą odwzorowania, oznaczając te stosunki na osi rzędnych i odpowiadające im stężenia alkoholu w mg na mililitr na osi odciętych.
6.4. Oznaczanie
6.4.1. Wprowadzić strzykawką 10 ml roztworu próbki (6.1.6) i zmierzyć powierzchnie pików alkoholu benzylowego i 4-etoksyfenolu. Obliczyć stosunek powierzchni pików alkoholu benzylowego do powierzchni pików 4-etoksyfenolu. Powtórzyć ten proces z dalszymi porcjami po 10 ml roztworów próbki aż do otrzymania zgodnych wyników.
6.4.2. Z krzywej odwzorowania (6.3.2) odczytać stężenie alkoholu benzylowego odpowiednio do stosunku powierzchni pików alkoholu benzylowego do powierzchni pików 4-etoksyfenolu.
7. OBLICZENIE
Obliczyć zawartość alkoholu benzylowego w próbce w procentach masowych według wzoru:
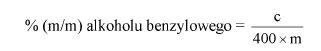
gdzie:
m = masa w gramach próbki pobranej do analizy (6.1.1),
c = stężenie alkoholu benzylowego w roztworze próbki (6.1.6) w mikrogramach na mililitr, otrzymane z krzywej odwzorowania.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości alkoholu benzylowego 1% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekroczyć 0,10%.
XXXIV. IDENTYFIKOWANIE CYRKONU I OZNACZANIE CYRKONU, GLINU I CHLORU W NIEAEROZOLOWYCH PRODUKTACH KOSMETYCZNYCH PRZECIWPOTOWYCH
Przedstawiono pięć procedur analitycznych:
A. Identyfikowanie cyrkonu
B. Oznaczanie cyrkonu
C. Oznaczanie glinu
D. Oznaczanie chloru
E. Obliczenie stosunków atomów glinu do atomów cyrkonu oraz sumy atomów glinu i cyrkonu do atomów chloru.
A. IDENTYFIKOWANIE CYRKONU
1. CEL I ZAKRES STOSOWANIA
Celem metody jest identyfikowanie cyrkonu w nieaerozolowych produktach kosmetycznych przeciwpotowych. Metoda nie jest odpowiednia do identyfikowania kompleksu glinowo-cyrkonowo-chlorowo-wodorotlenowego [AlxZr(OH)yClz nH2O].
2. ZASADA
Cyrkon identyfikuje się dzięki temu, że tworzy on z czerwienią alizarynową S charakterystyczny czerwono-fioletowy osad w środowisku silnie kwaśnym.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Kwas solny stężony, d20 = 1,18 g/ml.
3.2. Czerwień alizarynowa S (C.1.58005); 2% (m/v) wodny roztwór soli sodowej kwasu alizarynosulfonowego.
4. APARATURA
Zwykły sprzęt laboratoryjny.
5. PROCEDURA
5.1. Do ok. 1 g próbki w probówce dodać 2 ml wody. Zamknąć probówkę i wytrząsnąć.
5.2. Dodać trzy krople czerwieni alizarynowej S (3.2) i następnie 2 ml stężonego kwasu solnego (3.1), zamknąć i wytrząsnąć.
5.3. Pozostawić do odstania na ok. dwie minuty.
5.4. Czerwono-fioletowe zabarwienie cieczy i osadu znajdujące się na wierzchu wskazuje na obecność cyrkonu.
B. OZNACZANIE CYRKONU
1. CEL I ZAKRES STOSOWANIA
Metoda jest odpowiednia do oznaczania cyrkonu w kompleksach glinowo-cyrkonowo-chlorkowo-wodorotlenowych do maksymalnego stężenia 7,5% (m/m) cyrkonu w nieaerozolowych środkach przeciwpotowych.
2. ZASADA
Cyrkon ekstrahuje się z produktu w kwaśnym środowisku i oznacza metodą płomieniowej absorpcyjnej spektrometrii atomowej.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Kwas solny, stężony, d20 = 1,18 g/ml.
3.2. Kwas solny, roztwór 10% (v/v): do 500 ml wody w zlewce dodać 100 ml stężonego kwasu solnego (3.1), dokładnie wymieszać. Przenieść roztwór do jednolitrowej kolby miarowej i uzupełnić wodą do kreski.
3.3. Bazowy mianowany roztwór cyrkonu zawierający 1000 mg/ml w 0,5 M roztworze kwasu solnego (SpectrosoL lub równoważny).
3.4. Odczynnik 6-hydrat chlorku glinu [AlCl3 6H2O]: rozpuścić 22,6 g 6-hydratu chlorku glinu w 250 ml 10% (v/v) roztworu kwasu solnego (3.2).
3.5. Odczynnik chlorku amonu: rozpuścić 5,0 g chlorku amonu w 250 ml 10% (v/v) roztworu kwasu solnego (3.2).
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Mieszadło magnetyczne z ogrzewaniem.
4.3. Bibuła filtracyjna (Whatman nr 41 lub równoważna).
4.4. Spektrofotometr absorpcji atomowej wyposażony w lampę z katodą wnękową do oznaczania cyrkonu.
5. PROCEDURA
5.1. Przygotowanie próbki
5.1.1. Odważyć dokładnie ok. 1,0 g (m gramów) jednorodnej próbki produktu w 150 ml zlewce. Dodać 40 ml wody i 10 ml kwasu solnego (3.1).
5.1.2. Umieścić zlewkę w mieszadle magnetycznym z ogrzewaniem (4.2). Rozpocząć mieszanie i ogrzewać do wrzenia. Umieścić szkiełko zegarkowe na zlewce, aby zapobiec szybkiemu wysychaniu. Ogrzewać do wrzenia w ciągu pięciu minut, zdjąć zlewkę z mieszadła i schłodzić do temperatury pokojowej.
5.1.3. Przesączyć zawartość zlewki przez bibułę filtracyjną (4.3) do 100 ml kolby miarowej. Przepłukać zlewkę dwoma 10 ml porcjami wody i dodać przemywki po przesączeniu do kolby miarowej. Uzupełnić objętość wodą do kreski i wymieszać. Tego roztworu używa się również do oznaczania glinu (dział C).
5.1.4. Przenieść pipetą 20,00 ml roztworu próbki (5.1.3), 5,00 ml odczynnika chlorku glinu (3.4) i 5,00 ml odczynnika chlorku amonu (3.5) do 50 ml kolby miarowej. Uzupełnić objętość 10% (v/v) roztworem kwasu solnego (3.2) do kreski i wymieszać.
5.2. Warunki analizy metodą absorpcyjnej spektrometrii atomowej:
płomień: podtlenek azotu / acetylen
długość fali: 360,1 nm
korekcja tła: nie stosuje się
własności płomienia: bogaty, dla uzyskania maksymalnej absorbancji konieczna będzie optymalizacja wysokości palnika i warunków spalania.
5.3. Kalibrowanie
5.3.1. Przenieść pipetą po 5,00, 10,00, 15,00, 20,00 i 25,00 ml bazowego roztworu mianowanego cyrkonu (3.3) do serii 50 ml kolb miarowych. Do wszystkich kolb miarowych dodać pipetą po 5,00 ml odczynnika chlorku glinu (3.4) i 5,00 ml odczynnika chlorku amonu (3.5). Uzupełnić objętość 10% (v/v) roztworem kwasu solnego (3.2) do kreski i wymieszać. Te roztwory zawierają odpowiednio po 100, 200, 300, 400 i 500 mg cyrkonu w mililitrze. Podobnie przygotować roztwór ślepej próby niezawierający roztworu mianowanego cyrkonu.
5.3.2. Zmierzyć absorbancję roztworu ślepej próby (5.3.1) i stosować uzyskaną wartość jako odpowiadającą zerowemu stężeniu cyrkonu dla krzywej odwzorowania. Zmierzyć absorbancję wszystkich mianowanych roztworów cyrkonu do kalibrowania (5.3.1).
Wykreślić krzywą odwzorowania odpowiadającą stosunkom wartości absorbancji do stężenia cyrkonu.
5.4. Oznaczanie
Zmierzyć absorbancję roztworu próbki (5.1.4). Z krzywej odwzorowania odczytać stężenie cyrkonu odpowiadające wartości absorbancji otrzymanej dla roztworu próbki.
6. OBLICZENIE
Obliczyć zawartość cyrkonu w próbce w procentach masowych według wzoru:
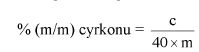
gdzie:
m = masa w gramach próbki pobranej do analizy (5.1.1),
c = stężenie cyrkonu w roztworze próbki (5.1.4), w mikrogramach na mililitr, otrzymane z kalibrowania.
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości cyrkonu 3,0% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,10% (m/m).
8. UWAGA
Zastosowanie metody plazmy sprzężonej indukcyjnie – emisyjnej spektrometrii atomowej jest dozwolone jako metody alternatywnej w stosunku do płomieniowej absorpcyjnej spektrometrii atomowej.
C. OZNACZANIE GLINU
1. CEL I ZAKRES STOSOWANIA
Celem metody jest oznaczanie glinu znajdującego się w kompleksach glinowo-cyrkonowo-chlorkowo-wodorotlenowych do maksymalnego stężenia 12% (m/m) glinu w nieaerozolowych środkach przeciwpotowych.
2. ZASADA
Glin ekstrahuje się z produktu w środowisku kwaśnym i oznacza metodą absorpcyjnej spektrometrii atomowej.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Kwas solny, stężony, d20 =1,18 g/ml.
3.2. Kwas solny, 1% (v/v): do 50 ml wody w zlewce dodać 10 ml stężonego kwasu solnego (3.1), dokładnie wymieszać. Przenieść roztwór do jednolitrowej kolby miarowej i uzupełnić objętość wodą do kreski.
3.3. Bazowy mianowany roztwór glinu zawierający 1000 mg/ml w 0,5 M roztworze kwasu azotowego (V) (SpectrosoL lub równoważny).
3.4. Odczynnik chlorku potasu: rozpuścić 10,0 g chlorku potasu w 250 ml 1% (v/v) roztworu kwasu solnego (3.2).
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Spektrometr absorpcji atomowej wyposażony w lampę z katodą wnękową do oznaczania glinu.
5. PROCEDURA
5.1. Przygotowanie próbki
Do oznaczania zawartości glinu używa się roztworu przygotowanego jak w opisie w B.5.1.3.
5.1.1. Przenieść pipetą 5,00 ml roztworu próbki (B.5.1.3) i 10,00 ml odczynnika chlorku potasu (3.4) do 100 ml kolby miarowej. Uzupełnić objętość 1% (v/v) roztworem kwasu solnego (3.2) do kreski i wymieszać.
5.2. Warunki analizy metodą absorbcyjnej spektrometrii atomowej:
płomień: podtlenek azotu / acetylen
długość fali: 309,3 nm
korekcja tła: nie stosuje się
własności płomienia: bogaty, dla uzyskania maksymalnej absorbancji konieczna jest optymalizacja wysokości palnika i warunków spalania.
5.3. Kalibrowanie
5.3.1. Przenieść pipetą po 1,00, 2,00, 3,00, 4,00 i 5,00 ml bazowego mianowanego roztworu glinu (3.3) do serii 100 ml kolb miarowych. Do wszystkich kolb miarowych przenieść pipetą po 1,00 ml odczynnika chlorku potasu (3.4) i uzupełnić objętość 1% (v/v) roztworem kwasu solnego (3.2) i wymieszać. Te roztwory zawierają po 10, 20, 30, 40 i 50 mg glinu w mililitrze.
Podobnie przygotować roztwór ślepej próby niezawierający roztworu mianowanego glinu.
5.3.2. Zmierzyć absorbancję roztworu ślepej próby (5.3.1) i stosować uzyskaną wartość jako odpowiadającą zerowemu stężeniu glinu dla krzywej odwzorowania. Zmierzyć absorbancję wszystkich mianowanych roztworów glinu do kalibrowania. Wykreślić krzywą odwzorowania odpowiadającą stosunkom wartości absorbancji do zawartości glinu.
5.4. Oznaczenie
Zmierzyć absorbancję roztworu próbki (5.1.1). Z krzywej odwzorowania odczytać stężenie glinu odpowiadające wartości absorbancji otrzymanej dla roztworu próbki.
6. OBLICZENIE
Obliczyć zawartość glinu w próbce w procentach masowych według wzoru:
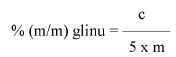
gdzie:
m = masa w gramach próbki pobranej do analizy (B.5.1.1),
c = stężenie glinu w roztworze próbki (5.1.1), w mikrogramach na mililitr, otrzymane z krzywej odwzorowania.
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości glinu 3,5% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,10% (m/m).
8. UWAGA
Zastosowanie metody plazmy sprzężonej indukcyjnie – emisyjnej spektrometrii atomowej jest dozwolone jako metody alternatywnej w stosunku do płomieniowej absorpcyjnej spektrometrii atomowej.
D. OZNACZANIE CHLORU
1. CEL I ZAKRES STOSOWANIA
Celem metody jest oznaczanie chloru obecnego w postaci jonu chlorkowego w kompleksie glinowo-cyrkonowo-wodorotlenowym w nieaerozolowych środkach przeciwpotowych.
2. ZASADA
Jon chlorkowy w produkcie oznacza się przez miareczkowanie potencjometryczne mianowanym roztworem azotanu (V) srebra.
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Kwas azotowy (V) stężony, d20 = 1,42 g/ml.
3.2. Kwas azotowy (V), 5% (v/v) roztwór: do 250 ml wody w zlewce dodać 25 ml stężonego kwasu azotowego (V) (3.1), starannie wymieszać. Przenieść ten roztwór do 500 ml kolby miarowej, uzupełnić wodą do kreski.
3.3. Aceton.
3.4. Azotan (V) srebra, 0,1 M roztwór mianowany (AnalaR lub równoważny).
4. APARATURA
4.1. Zwykły sprzęt laboratoryjny.
4.2. Mieszadło magnetyczne z ogrzewaniem.
4.3. Elektroda srebrna.
4.4. Kalomelowa elektroda odniesienia.
4.5. Miernik pH / miliwolt odpowiedni do miareczkowania potencjometrycznego.
5. PROCEDURA
5.1. Przygotowanie próbki
5.1.1. W 250 ml zlewce odważyć dokładnie ok. 1,0 g (m gramów) jednorodnej próbki produktu. Dodać 80 ml wody i 20 ml 5% (v/v) roztworu kwasu azotowego (V) (3.2).
5.1.2. Umieścić zlewkę na ogrzewanym mieszadle magnetycznym (4.2). Rozpocząć mieszanie i ogrzewać do wrzenia. Umieścić szkiełko zegarkowe na wierzchu zlewki, aby zapobiec szybkiemu wysychaniu. Ogrzewać do wrzenia w ciągu pięciu minut, zdjąć zlewkę z ogrzewacza i schłodzić do temperatury pokojowej.
5.1.3. Dodać 10 ml acetonu (3.3), zanurzyć elektrody (4.3 i 4.4) poniżej powierzchni roztworu i rozpocząć mieszanie.
Miareczkować potencjometrycznie 0,1 M roztworem azotanu (V) srebra (3.4) i wykreślić krzywą różniczkową do oznaczania punktu końcowego (V ml).
6. OBLICZENIE
Obliczyć zawartość chloru w próbce w procentach masowych według wzoru:
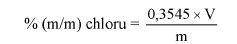
gdzie:
m = masa próbki pobranej do analizy (5.1.1) w gramach,
V = objętość 0,1 M roztworu azotanu (V) srebra, w mililitrach, zużytego do miareczkowania w punkcie końcowym (5.1.3).
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości chloru 4% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,10% (m/m).
E. OBLICZENIE STOSUNKU ATOMÓW GLINU DO ATOMÓW CYRKONU ORAZ SUMY ATOMÓW GLINU I CYRKONU DO ATOMÓW CHLORU
1. Obliczenie stosunku atomów glinu do atomów cyrkonu
Obliczyć stosunek Al : Zr według wzoru:
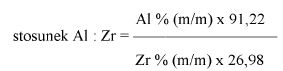
2. Obliczenie stosunku sumy atomów glinu i cyrkonu do atomów chloru
Obliczyć stosunek (Al + Zr) : Cl według wzoru:

XXXV. IDENTYFIKOWANIE I OZNACZANIE HEKSAMIDYNY, DIBROMO-HEKSAMIDYNY, DIBROMOPROPAMIDYNY I CHLOROHEKSYDYNY
1. CEL I ZAKRES STOSOWANIA
Celem metody jest jakościowe i ilościowe oznaczanie:
1) heksamidyny i jej soli, łącznie z izetionianem i 4-hydroksybenzoesanem;
2) dibromoheksamidyny i jej soli, łącznie z izetionianem;
3) dibromopropamidyny i jej soli, łącznie z izetionianem;
4) dioctanu, diglukonianu i chlorowodorku chloroheksydyny w produktach kosmetycznych.
2. DEFINICJA
Stężenia heksamidyny, dibromoheksamidyny, dibromopropamidyny i chloroheksydyny oznaczone tą metodą są wyrażone jako procent masowy (% m/m).
3. ZASADA
Identyfikowanie i oznaczanie wykonuje się metodą wysokosprawnej chromatografii cieczowej (HPLC) na fazie odwróconej, jonów dwubiegowych z następną detekcją spektrometryczną w zakresie nadfioletu. Heksamidynę, dibromoheksamidynę, dibromopropamidynę i chloroheksydynę identyfikuje się na podstawie ich czasu retencji po rozdziale na kolumnie chromatograficznej.
4. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
4.1. Metanol.
4.2. Monohydrat heptano-1-sulfonian sodu.
4.3. Kwas octowy lodowaty, d20 = 1,05 g/ml.
4.4. Chlorek sodowy.
4.5. Fazy ruchome
4.5.1. Rozpuszczalnik I: 0,005 M roztwór monohydratu i heptano-1-sulfonianu sodu (4.2) w metanolu nastawiony na pH 3,5 kwasem octowym lodowatym (4.3).
4.5.2. Rozpuszczalniki II: 0,005 M roztwór monohydratu heptano-1-sulfonianu sodu (4.2) w wodzie nastawiony na pH 3,5 kwasem octowym lodowatym (4.3).
Uwaga: Jeżeli jest konieczne poprawienie kształtu pików, można zmodyfikować fazy ruchome i przygotować je następująco:
1) rozpuszczalnik I: rozpuścić 5,84 g chlorku sodowego (4.4) i 1,1013 g monohydratu i heptano-1-sulfonianu sodu (4.2) w 100 ml wody, dodać 900 ml metanolu (4.1) i nastawić na wyraźne pH 3,5 kwasem octowym lodowatym (4.3);
2) rozpuszczalnik II: rozpuścić 5,84 g chlorku sodowego (4.4) i 1,1013 g monohydratu 1-heptanosulfonianu sodu (4.2) w jednym litrze wody i nastawić na pH 3,5 kwasem octowym lodowatym (4.3).
4.6. Diizetionan heksamidyny [C20H24N4O2 2C2H6O4S].
4.7. Diizetionian dibromoheksamidyny [C20H24Br2N4O2 2C2H4O4S].
4.8. Diizetionian dibromopropamidyny [C17H18Br2N4O2O 2C2H6O4S].
4.9. Dioctan chloroheksydyny [C22H30Cl2N10 2C2H4O2].
4.10. Roztwory odniesienia: przygotować 0,05% (m/v) roztwory wszystkich czterech środków konserwujących (4.6.–4.9) w rozpuszczalniku I (4.5.1).
4.11. 3,4,4'-trichlorokarbanilid (triclocarban).
4.12. 4,4'-dichloro-3-(trifluorometylo)-karbanilid (halocarban).
5. APARATURA
5.1. Zwykły sprzęt laboratoryjny.
5.2. Wysokosprawny chromatograf cieczowy z detektorem w zakresie promieniowania nadfioletowego o zmiennej długości fali.
5.3. Kolumna analityczna: stal kwasoodporna, długość 30 cm, średnica wewnętrzna 4 mm, z wypełnieniem µ-Bondapack Cg, 10 µm lub równoważnym.
5.4. Łaźnia ultradźwiękowa.
6. IDENTYFIKOWANIE
6.1. Przygotowanie próbki
Odważyć ok. 0,5 g próbki w 10 ml kolbie miarowej i uzupełnić objętość do kreski rozpuszczalnikiem I (4.5.1). Umieścić kolbę w łaźni ultradźwiękowej (5.4) na 10 minut. Przesączyć lub odwirować roztwór. Zebrać przesącz lub ciecz znajdującą się na wierzchu do analizy chromatograficznej.
6.2. Chromatografia
6.2.1. Gradientowy przepływ fazy ruchomej
| Czas, w min | Rozpuszczalnik I, w % v/v (4.5.1) | Rozpuszczalnik II, w % v/v (4.5.2) |
| 0 | 50 | 50 |
| 15 | 65 | 35 |
| 30 | 65 | 35 |
| 45 | 50 | 50 |
6.2.2. Nastawić szybkość przepływu fazy ruchomej (6.2.1) na 1,5 ml/min, a temperaturę kolumny na temperaturę 35°C.
6.2.3. Nastawić długość fali detektora na 264 nm.
6.2.4. Wprowadzić strzykawką po 10 µl wszystkich roztworów odniesienia (4.10) i zarejestrować ich chromatogramy.
6.2.5. Wprowadzić strzykawką 10 µl roztworu próbki (6.1) i zarejestrować jego chromatogram.
6.3. Zidentyfikować, czy w próbce znajduje się heksamidyna, dibromoheksamidyna, dibromopropamidyna lub chloroheksydyna przez porównanie czasu retencji pików zarejestrowanych zgodnie z pkt 6.2.5 z czasem retencji otrzymanym dla roztworów odniesienia zgodnie z pkt 6.2.4.
7. OZNACZANIE
7.1. Oznaczanie
Przygotowanie roztworów odniesienia: Jako wewnętrzny standard zastosować jeden ze środków konserwujących (4.6–4.9) nieznajdujący się w próbce. Jeżeli nie jest to możliwe, można użyć triclocarbanu (4.11) lub halocarbanu (4.12).
7.1.1. Bazowy 0,05% (m/v) roztwór środka konserwującego zidentyfikowanego zgodnie z pkt 6.3 w rozpuszczalniku I.
7.1.2. Bazowy 0,05% (m/v) roztwór środka konserwującego wybranego jako wewnętrzny standard w rozpuszczalniku I.
7.1.3. Przygotować cztery mianowane roztwory dla wszystkich zidentyfikowanych środków konserwujących przez przeniesienie do serii 10 ml kolb miarowych objętości roztworów bazowych zidentyfikowanych środków konserwujących (7.1.1) i odpowiednich objętości bazowego wewnętrznego roztworu mianowanego (7.1.2) zgodnie z poniższą tabelą. Uzupełnić objętość we wszystkich kolbach rozpuszczalnikiem (4.5.1) do kreski i wymieszać.
| Mianowany roztwór | Bazowy wewnętrzny roztwór mianowany | Bazowy roztwór zidentyfikowanego środka konserwującego | |
| ilość dodanych ml (7.1.2) | ilość dodanych ml (7.1.1) | ml/ml* | |
| I | 1,0 | 0,5 | 25 |
| II | 1,0 | 1,0 | 50 |
| III | 1,0 | 1,5 | 75 |
| IV | 1,0 | 2,0 | 100 |
| * Wartości te podano jako wskazówkę i odpowiadają one stężeniom zidentyfikowanych środków konserwujących w roztworach mianowanych przygotowanych z użyciem roztworu bazowego, który zawiera dokładnie 0,05% zidentyfikowanego środka konserwującego. | |||
7.2. Przygotowanie próbki
7.2.1. W 10 ml kolbie miarowej odważyć dokładnie ok. 0,5 g (g gramów) próbki, dodać 1,0 ml wewnętrznego roztworu mianowanego (7.1.2) i 6 ml rozpuszczalnika (4.5.1) i wymieszać.
7.2.2. Umieścić kolbę w łaźni naddźwiękowej (5.4) na 10 minut. Schłodzić. Uzupełnić objętość rozpuszczalnikiem I do kreski i wymieszać. Odwirować lub przesączyć przez karbowany sączek z bibuły filtracyjnej. Zebrać ciecz znajdującą się na wierzchu lub przesącz zależnie od stosowanego sposobu analizy chromatograficznej.
7.3. Chromatografia
7.3.1. Nastawić gradient fazy ruchomej, szybkość przepływu fazy ruchomej, temperaturę kolumny i długość fali w detektorze UV w aparaturze HPLC (5.2) na warunki takie, jakie są wymagane na etapie identyfikowania (6.2.1.–6.2.3).
7.3.2. Wprowadzić strzykawką 10 µl roztworu próbki (7.2.2) i zmierzyć powierzchnię pików. Powtórzyć ten proces z dalszymi 10 µl porcjami roztworu próbki aż do uzyskania zgodnych wyników. Obliczyć stosunek powierzchni piku odpowiadającego analizowanemu związkowi do powierzchni piku odpowiadającego wewnętrznemu standardowi.
7.4. Kalibrowanie
7.4.1. Wprowadzić strzykawką po 10 µl roztworów mianowanych (7.1.3) i zmierzyć powierzchnię pików.
7.4.2. Dla każdego roztworu mianowanego (7.1.3) obliczyć stosunek powierzchni piku heksamidyny, dibromoheksamidyny, dibromopropamidyny lub chloroheksydyny do powierzchni piku wewnętrznego standardu. Wykreślić krzywą odwzorowania, oznaczając te stosunki na osi rzędnych i odpowiednie stężenia zidentyfikowanych środków konserwujących w roztworach mianowanych, w mikrogramach na mililitr, na osi odciętych.
7.4.3. Z krzywej odwzorowania (7.4.2) odczytać stężenie zidentyfikowanego środka konserwującego odpowiadające stosunkowi powierzchni piku obliczonemu jak w pkt 7.3.2.
8. OBLICZENIE
Obliczyć zawartość heksamidyny, dibromoheksamidyny, dibromopropamidyny lub chloroheksydyny w próbce w procentach masowych według wzoru:
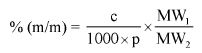
gdzie:
p = masa próbki pobranej do analizy, w gramach (7.2.1),
c = stężenie środka konserwującego w roztworze próbki, w mikrogramach na mililitr, otrzymane z krzywej odwzorowania,
MW1 = ciężar cząsteczkowy podstawowej formy obecnego środka konserwującego,
MW2 = ciężar cząsteczkowy odpowiedniej soli (10).
9. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla stężenia heksamidyny, dibromoheksamidyny dibromopropamidyny lub chloroheksydyny 0,1% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki, nie powinna przekraczać 0,005%.
10. TABLICA CIĘŻARÓW CZĄSTECZKOWYCH
| heksamidyna | C20H26N4O2 | 354,45 |
| 2-hydroksyetanosulfonian | C20H26N4O2 2C2H6O4S | 606,72 |
| heksamidyny |
|
|
| di-p-hydroksybenzoesan | C20H26N4O2 2C7H6O3 | 630,71 |
| heksamidyny |
|
|
| dibromoheksamidyna | C20H24Br2N4O2 | 512,24 |
| 2-hydroksyetanosulfonian | C20H24Br2N4O2 2C2H6O4S | 764,51 |
| dibromoheksamidyny |
|
|
| dibromopropamidyna | C17H18Br2N4O2 | 470,18 |
| 2-hydroksyetanosulfonian | C17H18Br2N4O2 2C2H6O4S | 722,43 |
| dibromopropamidyny |
|
|
| chlorheksydyna | C22H30Cl2N10 | 505,45 |
| dioctan chlorheksydyny | C22H30Cl2N10 2C2H4O2 | 625,56 |
| diglukonian chlorheksydyny | C2H30Cl2N10 2C6H12O7 | 897,76 |
| dichlorowodorek chlorheksydyny | C22H30Cl2N10 2HCl | 578,37 |
XXXVI. IDENTYFIKOWANIE I OZNACZANIE KWASU BENZOESOWEGO, KWASU 4-HYDROKSYBENZOESOWEGO, KWASU SORBINOWEGO, KWASU SALICYLOWEGO I KWASU PROPIONOWEGO W PRODUKTACH KOSMETYCZNYCH
1. CEL I ZAKRES ZASTOSOWANIA
Metoda jest stosowana do identyfikowania i oznaczania kwasu benzoesowego, kwasu 4-hydroksybenzoesowego, kwasu sorbinowego, kwasu salicylowego i kwasu propionowego w produktach kosmetycznych. Oddzielne procedury dotyczą identyfikowania tych środków konserwujących, a w części B podano procedury oznaczania kwasu propionowego i oznaczanie kwasu 4-hydroksybenzoesowego, kwasu salicylowego, kwasu sorbinowego i kwasu benzoesowego.
2. DEFINICJA
Ilość kwasu benzoesowego, kwasu 4-hydroksybenzoesowego, kwasu salicylowego, kwasu sorbinowego i kwasu propionowego oznaczona tą metodą jest wyrażona w procentach masowych wolnych kwasów.
A. IDENTYFIKOWANIE
1. ZASADA
Ekstrakt otrzymany przez kwasowo-zasadową ekstrakcję środków konserwujących analizuje się metodą chromatografii cienkowarstwowej (TLC) w ciągu jednego dnia po otrzymaniu pochodnej. Zależnie od wyników identyfikacja zostaje potwierdzona metodą wysokosprawnej chromatografii cieczowej (HPLC) lub w przypadku kwasu propionowego metodą chromatografii gazowej (GC).
2. ODCZYNNIKI
2.1. Zasada ogólna
Wszystkie odczynniki muszą być czyste do analizy. Używana woda musi być wodą destylowaną lub wodą co najmniej równoważnej czystości.
2.2. Aceton.
2.3. Eter dietylowy.
2.4. Acetonitryl.
2.5. Toluen.
2.6. n-heksan.
2.7. Ciekła parafina.
2.8. Kwas solny, 4 M roztwór.
2.9. Wodorotlenek potasu, 4 M roztwór wodny.
2.10. 2-hydrat chlorku wapnia, CaCl2 2H2O.
2.11. Węglan litu, Li2CO3.
2.12. 2-bromo-2'acetonaftyl.
2.13. Kwas 4-hydroksybenzoesowy.
2.14. Kwas salicylowy.
2.15. Kwas benzoesowy.
2.16. Kwas sorbinowy.
2.17. Kwas propionowy.
2.18. Roztwory odniesienia
Przygotować 0,1% (m/v) roztwory (100 mg / 100 ml) wszystkich pięciu środków konserwujących (2.13. – do 2.17) w eterze dietylowym.
2.19. Odczynnik do otrzymywania pochodnych
0,5% (m/v) roztwór 2-bromo-2'acetonaftyl (2.12) w acetonitrylu (2.4) (50 mg/10 ml). Roztwór ten powinien być przygotowywany co tydzień i przechowywany w lodówce.
2.20. Roztwór katalizatora
0,3% (m/v) roztwór węglanu litu (2.11) w wodzie (300 mg/100 ml). Roztwór powinien być świeżo przygotowany.
2.21. Rozpuszczalnik rozwijający
Toluen (2.5) / aceton (2.2) (20:0,5, v/v)
2.22. Ciekła parafina (2.7) / n-heksan (2.6) (1:2, v/v).
3. APARATURA
Zwykły sprzęt laboratoryjny.
3.1. Łaźnia wodna z możliwością utrzymania temperatury 60°C.
3.2. Komora do rozwijania chromatogramu.
3.3. Źródło światła ultrafioletowego, 254 i 366 nm.
3.4. Płytki cienkowarstwowe, Kieselgel 60, bez wskaźnika fluorescencji, 20 x 20 cm, grubość warstwy 0,25 mm ze strefą nanoszenia 2,5 x 20 cm (Merck 11845 lub równoważne).
3.5. Mikrostrzykawka, 10 µl.
3.6. Mikrostrzykawka, 25 µl.
3.7. Suszarka z możliwością utrzymania temperatury do temperatury 105°C.
3.8. 50 ml szklane rurki z gwintowanym korkiem.
3.9. Bibuła filtracyjna, średnica 90 mm (Schleicher and Schüll, Weissband nr 5892 lub równoważna).
3.10. Uniwersalny papierek wskaźnikowy pH = 1–11.
3.11. 5 ml szklane fiolki na próbki.
3.12. Rotacyjna wyparka cienkowarstwowa (Rotavapor lub równoważna).
3.13. Ogrzewana płytka.
4. PROCEDURA
4.1. Przygotowanie próbki
W 50 ml szklanej rurce z gwintowanym korkiem (3.8) odważyć ok. 1 g próbki. Dodać cztery krople 4 M kwasu solnego (2.8) i 40 ml acetonu (2.2). Dla silnie alkalicznych wyrobów, takich jak mydło toaletowe, należy dodać 20 kropli 4M kwasu solnego.
Papierkiem wskaźnikowym (3.10) sprawdzić, czy pH wynosi ok. 2. Zamknąć rurkę i wytrząsać energicznie w ciągu jednej minuty.
Jeżeli konieczne jest ułatwienie ekstrakcji środka konserwującego do fazy acetonowej, należy ogrzewać łagodnie mieszaninę do temperatury ok. 60°C do stopienia fazy ciekłej.
Schłodzić roztwór do temperatury pokojowej i przesączyć przez bibułę filtracyjną (3.9) do kolby stożkowej. Przenieść 20 ml przesączu do 200 ml kolby stożkowej, dodać 20 ml wody i wymieszać. Dostosować odczyn mieszaniny do pH ok. 10 wodorotlenkiem potasu 4M (2.9), używając papierka wskaźnikowego (3.10) do oznaczenia pH.
Dodać 1 g chlorku wapnia (2.10) i energicznie wytrząsnąć. Przesączyć przez bibułę filtracyjną (3.9) do 250 ml lejka rozdzielającego zawierającego 75 ml eteru dietylowego (2.3) i wytrząsać energicznie w ciągu jednej minuty. Pozostawić do rozdzielenia i przenieść warstwę wodną do 250 ml kolby stożkowej. Odrzucić warstwę eterową. Używając papierka wskaźnikowego (3.10), doprowadzić odczyn do pH ok. 2 kwasem solnym 4 M (2.8). Dodać 10 ml eteru dietylowego (2.3), zamknąć kolbę i wytrząsać energicznie w ciągu jednej minuty, pozostawić do rozdzielenia i przenieść warstwę eterową do rotacyjnej wyparki filmowej (3.12). Odrzucić warstwę wodną.
Odparować warstwę eterową prawie do sucha i ponownie rozpuścić pozostałość w 1 ml eteru dietylowego (2.3). Przenieść roztwór do fiolki na próbkę (3.11).
4.2. Chromatografia cienkowarstwowa
Dla wszystkich roztworów odniesienia i próbek analizowanych chromatograficznie nanieść strzykawką (3.5) po ok. 3 µl węglanu litu (2.20) w równych odległościach na linii początkowej w strefie nanoszenia płytki do chromatografii cienkowarstwowej (3.4) i wysuszyć w strumieniu zimnego powietrza.
Umieścić płytkę cienkowarstwową na ogrzewanej płytce (3.13) podgrzanej do temperatury 40°C dla otrzymania możliwie najmniejszych plam. Nanieść mikrostrzykawką (3.5) po 10 µl wszystkich roztworów odniesienia (2.18) i roztworu próbki (4.1) na linii początkowej płytki, dokładnie w miejscach plam, w których naniesiono roztwór węglanu litu.
Nanieść ponownie po ok. 15 µl odczynnika do otrzymywania pochodnych (2.19) (roztworu 2-acetylo-7-bromonaftalenu), dokładnie w miejscach plam, w których były naniesione roztwory odniesienia i próbki oraz roztwór węglanu litu.
Ogrzewać płytkę do chromatografii cienkowarstwowej w suszarce (3.7) w temperaturze 80°C w ciągu 45 minut. Po schłodzeniu rozwijać płytkę w komorze (3.2), którą doprowadzono do stanu równowagi w ciągu 15 minut (bez wyłożenia wnętrza bibułą filtracyjną), używając rozpuszczalnika rozwijającego (2.21) (toluen/aceton), aż do osiągnięcia przez czoło rozpuszczalnika odległości 15 cm (może to trwać ok. 80 minut).
Wysuszyć płytkę w strumieniu zimnego powietrza i oceniać otrzymane plamy w świetle UV (3.3). Dla wzmocnienia fluorescencji słabo widocznych plam można zamoczyć płytkę do chromatografii cienkowarstwowej w mieszaninie ciekłej parafina/n-heksan (2.22).
5. IDENTYFIKACJA
Obliczyć współczynnik Rf dla wszystkich plam. Porównać wartość Rf i zachowanie próbki pod wpływem promieniowania UV z wartościami otrzymanymi dla roztworów odniesienia. Sformułować wstępny wniosek o obecności i zidentyfikowaniu środka konserwującego znajdującego się w próbce. Przeprowadzić analizę metodą HPLC opisaną w części B lub, jeżeli jest podejrzewana obecność kwasu propionowego w próbce, analizę metodą GC opisaną w części C. Porównać czas retencji z czasem otrzymanym dla roztworów odniesienia.
Podsumować wyniki otrzymane metodami TLC i HPLC lub GC oraz potwierdzić ostateczną identyfikację środków konserwujących znajdujących się w próbce na podstawie otrzymanych wyników.
B. OZNACZANIE KWASU BENZOESOWEGO, KWASU 4-HYDROKSYBENZOESOWEGO, KWASU SORBINOWEGO I KWASU SALICYLOWEGO
1. ZASADA
Próbkę po zakwaszeniu ekstrahuje się mieszaniną alkoholu etylowego i wody. Po przesączeniu środki konserwujące oznacza się metodą wysokosprawnej chromatografii cieczowej (HPLC).
2. ODCZYNNIKI
2.1. Wszystkie odczynniki muszą być czyste do analizy i jeżeli potrzeba – odpowiednie do HPLC. Używana woda musi być wodą destylowaną lub co najmniej równoważnej czystości.
2.2. Alkohol etylowy, absolutny.
2.3. Kwas 4-hydroksybenzoesowy.
2.4. Kwas salicylowy.
2.5. Kwas benzoesowy.
2.6. Kwas sorbinowy.
2.7. Octan sodu (CH3COONa 3H2O).
2.8. Kwas octowy, (a)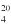
2.9. Acetonitryl.
2.10. Kwas siarkowy (VI), 2 M.
2.11. Wodorotlenek potasu, 0,2 M roztwór wodny.
2.12. Kwas 2-metoksybenzoesowy.
2.13. Mieszanina alkohol etylowy / woda
Zmieszać dziewięć objętości alkoholu etylowego (2.2) i jedną objętość wody (2.1).
2.14. Roztwór wewnętrznego standardu
Przygotować roztwór zawierający w przybliżeniu 1 g kwasu 2-metoksybenzoesowego (2.12) w 500 ml mieszaniny alkohol etylowy/woda (2.13).
2.15. Faza ruchoma do HPLC
2.15.1. Bufor octanowy: do 1 l wody dodać 6,35 g octanu sodu (2.7) i 20,0 ml kwasu octowego (2.8) i wymieszać.
2.15.2. Przygotować fazę ruchomą przez zmieszanie dziewięciu objętości buforu octanowego (2.15.1) i jednej objętości acetonitrylu (2.9).
2.16. Bazowy roztwór środków konserwujących
Odważyć dokładnie ok. 0,05 g kwasu 4-hydroksybenzoesowego (2.3), 0,2 g kwasu salicylowego (2.4), 0,2 g kwasu benzoesowego (2.5) i 0,05 g kwasu sorbinowego (2.6) w 50 ml kolbie miarowej i uzupełnić objętość do kreski mieszaniną alkohol etylowy / woda (2.13). Przechowywać przyrządzony roztwór w lodówce. Roztwór jest trwały w ciągu jednego tygodnia.
2.17. Mianowane roztwory środków konserwujących
Do serii 20 ml kolb miarowych przenieść odpowiednio po: 8,00; 4,00; 2,00; 1,00 i 0,50 ml bazowego roztworu środków konserwujących (2.16). Do wszystkich kolb dodać po 10,00 ml roztworu wewnętrznego standardu (2.14) i 0,5 ml 2 M kwasu siarkowego (VI) (2.10). Uzupełnić objętość do kreski mieszaniną alkohol etylowy / woda (2.13). Roztwory te muszą być świeżo przygotowane.
3. APARATURA
Zwykły sprzęt laboratoryjny.
3.1. Łaźnia wodna nastawiona na temperaturę 60°C.
3.2. Wysokosprawny chromatograf cieczowy z detektorem o zmiennej długości fali UV i pętlą 10 µl dla wprowadzenia próbki.
3.3. Kolumna analityczna
Stal kwasoodporna, długość 12,5 do 25 cm, średnica wewnętrzna 4,6 mm, z wypełnieniem Nucleosil 5C18 lub równoważnym.
3.4. Bibuła filtracyjna, średnica 90 mm, Schleicher and Schüll, Weissband nr 5892 lub równoważna.
3.5. 50 ml rurki szklane z gwintowanym korkiem.
3.6. 5 ml szklane fiolki na próbki.
3.7. Kamyczki wrzenne, o wymiarach 2 do 4 mm, z karborundu (węgliku krzemu) lub równoważne.
4. PROCEDURA
4.1. Przygotowanie próbki.
4.1.1. Przygotowanie próbki bez dodawania wewnętrznego standardu.
Odważyć 1 g próbki w 50 ml szklanej rurce z gwintowanym korkiem (3.5). Dodać pipetą do rurki 1,00 ml 2 M kwasu siarkowego (VI) (2.10) i 40 ml mieszaniny alkohol etylowy / woda (2.13). Dodać ok. 1 g kamyczków wrzennych (3.7), zamknąć rurkę i wytrząsać energicznie w ciągu co najmniej jednej minuty aż do otrzymania zawiesiny jednorodnej. Dla ułatwienia ekstrakcji środków konserwujących do fazy etanolowej należy umieścić rurkę w łaźni wodnej (3.1) utrzymywanej w temperaturze 60°C na okres dokładnie pięciu minut. Natychmiast schłodzić rurkę w strumieniu zimnej wody i przechować ekstrakt w temperaturze 5°C w ciągu jednej godziny. Przesączyć ekstrakt przez bibułę filtracyjną (3.4). Przenieść ok. 2 ml ekstraktu do fiolki na próbkę (3.6). Przechowywać ekstrakt w temperaturze 5°C i wykonać oznaczenie metodą HPLC w ciągu 24 godzin od przygotowania.
4.1.2. Przygotowanie próbki z dodawaniem wewnętrznego standardu
Odważyć z dokładnością do trzeciego miejsca po przecinku 1 +/–0,1 g (a gramów) próbki w 50 ml rurce szklanej z gwintowanym korkiem (3.5). Dodać pipetą 1,00 ml 2 M kwasu siarkowego (VI) (2.10) i 30 ml mieszaniny alkohol etylowy / woda (2.13). Dodać ok. 1 g kamyczków wrzennych do ogrzewania do wrzenia (3.7) i 10,00 ml roztworu standardu wewnętrznego (2.14). Zamknąć rurkę i wytrząsać energicznie w ciągu co najmniej jednej minuty do otrzymania zawiesiny jednorodnej. Dla ułatwienia ekstrakcji środków konserwujących do fazy etanolowej należy umieścić rurkę w łaźni wodnej (3.1) utrzymywanej w temperaturze 60°C na okres dokładnie pięciu minut.
Schłodzić natychmiast rurkę w strumieniu zimnej wody i przechowywać ekstrakt w temperaturze 5°C w ciągu jednej godziny.
Przesączyć ekstrakt przez bibułę filtracyjną (3.4). Przenieść ok. 2 ml przesączu do fiolki na próbkę (3.6). Przechowywać przesącz w temperaturze 5°C i wykonać oznaczenie metodą HPLC w ciągu 24 godzin od przygotowania.
4.2. Wysokosprawna chromatografia cieczowa
Faza ruchoma: acetonitryl / bufor octanowy (2.15).
Nastawić szybkość przepływu fazy ruchomej przez kolumnę na 2,0 +/– 0,5 ml/minutę.
Ustawić długość fali w detektorze na 240 nm.
4.2.1. Kalibrowanie
Wprowadzić strzykawką 10 µl porcje kolejno wszystkich roztworów mianowanych środków konserwujących (2.17) do chromatografu cieczowego (3.2). Dla wszystkich roztworów oznaczyć stosunki wysokości pików analizowanych środków konserwujących do wysokości piku wewnętrznego standardu otrzymane z chromatogramów. Sporządzić dla wszystkich środków konserwujących wykres zależności stosunku wysokości pików od stężenia. Upewnić się, że podczas kalibrowania otrzymuje się liniową zależność dla mianowanych roztworów.
4.2.2. Oznaczanie
Do chromatografu cieczowego (3.2) wprowadzić strzykawką 10 µl ekstraktu próbki (4.1.1) i zarejestrować chromatogram. Wprowadzić strzykawką 10 µl roztworu środków konserwujących i zarejestrować chromatogram. Porównać otrzymane chromatogramy. Jeżeli na chromatogramie ekstraktu próbki (4.1.1) nie występuje pik mający w przybliżeniu ten sam czas retencji jak kwas 2-metoksybenzoesowy (polecany jako standard wewnętrzny), wprowadzić strzykawką 10 µl ekstraktu próbki z dodanym standardem wewnętrznym (4.1.2) do chromatografu cieczowego i zarejestrować chromatogram.
Jeżeli obserwuje się na chromatogramie ekstraktu próbki (4.1.1) pik przeszkadzający o takim samym czasie retencji jak kwas 2-metoksybenzoesowy, należy wybrać inny odpowiedni standard wewnętrzny (jeżeli jeden z badanych środków konserwujących nie występuje na chromatogramie, ten środek może być używany jako wewnętrzny standard).
Upewnić się, czy chromatogramy otrzymane dla roztworu wewnętrznego standardu i roztworu próbki odpowiadają następującym wymaganiom:
1) Rozdział pików w najgorszej rozdzielonej parze wynosi najmniej 0,90 (definicje rozdziału pików przedstawiono na rys. 13).
Rys. 13
Rozdział pików
Jeżeli nie uzyskano wymaganego rozdziału pików, powinno się zastosować kolumnę o większej zdolności rozdzielczej lub poprawić skład fazy ruchomej aż do spełnienia wymagań.
2) Współczynnik asymetrii As wszystkich otrzymanych pików znajduje się w zakresie między 0,9 i 1,5 (definicję współczynników asymetrii piku przedstawiono na rys. 14); do zarejestrowania chromatogramu w celu oznaczenia współczynnika asymetrii polecana jest szybkość przesuwu papieru w rejestratorze co najmniej 2 cm/minutę.
Rys. 14
Współczynnik asymetrii piku
3) Otrzymuje się stałą linię bazową.
5. OBLICZENIE
Do obliczenia stężenia kwasowych środków konserwujących w roztworze próbki należy stosować stosunki wysokości pików badanych środków konserwujących do wysokości piku kwasu 2-metoksybenzoesowego (wewnętrzny standard) i wykres kalibracyjny.
Zawartość w próbce kwasu benzoesowego, kwasu 4-hydroksybenzoesowego, kwasu sorbinowego lub kwasu salicylowego obliczyć w procentach masowych (xi) według wzoru:
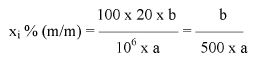
gdzie:
a = masa w gramach próbki pobranej do analizy (4.1.2),
b = stężenie środka konserwującego (µg/ml) w ekstrakcie próbki (4.1.2) otrzymane z wykresu kalibracyjnego.
6. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości kwasu 4-hydroksybenzoesowego 0,40% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,035%.
Dla zawartości kwasu benzoesowego 0,50% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,050%.
Dla zawartości kwasu salicylowego 0,50% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,045%.
Dla zawartości kwasu sorbinowego 0,60% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,035%.
7. UWAGI
7.1. Wyniki testu odchyleń wykonane dla tej metody wykazały, że ilość kwasu siarkowego (VI) dodanego do ekstraktu kwasów z próbki jest krytyczna i ustalone ograniczone ilości próbki powinny być utrzymane w zalecanych granicach.
7.2. Jeżeli potrzeba, można stosować odpowiednią kolumnę zabezpieczającą.
C. OZNACZANIE KWASU PROPIONOWEGO
1. CEL I ZAKRES
Metoda jest odpowiednia do oznaczania kwasu propionowego o maksymalnym stężeniu 2% (m/m) w produktach kosmetycznych.
2. DEFINICJA
Stężenie kwasu propionowego określone tą metodą jest wyrażone w procentach masowych (% m/m) wyrobu.
3. ZASADA
Po ekstrakcji kwasu propionowego z wyrobu oznaczenie przeprowadza się metodą chromatografii gazowej z użyciem kwasu 2-metylopropionowego jako wewnętrznego standardu.
4. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy; musi być używana woda destylowana lub woda o równoważnej jakości.
4.1. Alkohol etylowy 96% (v/v).
4.2. Kwas propionowy.
4.3. Kwas 2-metylopropionowy.
4.4. Kwas ortofosforowy (VI), 10% (m/v).
4.5. Roztwór kwasu propionowego.
Odważyć dokładnie ok. 1,00 g (p gramów) kwasu propionowego w 50 ml kolbie miarowej i uzupełnić objętość alkoholem etylowym (4.1).
4.6. Roztwór wewnętrznego standardu
Odważyć dokładnie ok. 1,00 (e gramów) kwasu 2-metylopropionowego w 50 ml kolbie miarowej i uzupełnić objętość alkoholem etylowym (4.1).
5. APARATURA
5.1. Zwykły sprzęt laboratoryjny.
5.2. Chromatograf gazowy z detektorem płomieniowo-jonizacyjnym.
5.3. Szklana rurka (20 x 150 mm) z korkiem gwintowanym.
5.4. Łaźnia wodna o temperaturze 60°C.
5.5. 10 ml szklana strzykawka z membraną filtracyjną o średnicy porów 0,45 µm.
6. PROCEDURA
6.1. Przygotowanie próbki
6.1.1. Przygotowanie próbki bez wewnętrznego standardu.
W szklanej rurce (5.3) odważyć ok. 1 g próbki. Dodać 0,5 ml kwasu fosforowego (V) (4.4) i 9,5 ml alkoholu etylowego (4.1).
Zamknąć rurkę i energicznie wytrząsnąć. Jeżeli to konieczne, umieścić rurkę w łaźni wodnej ogrzewanej do temperatury 60°C (5.4) na pięć minut w celu całkowitego rozpuszczenia fazy lipidowej. Schłodzić szybko pod strumieniem bieżącej wody. Przesączyć część roztworu przez filtr membranowy (5.5). Zanalizować chromatograficznie przesącz w ciągu tego samego dnia.
6.1.2. Przygotowanie próbki z wewnętrznym standardem
Odważyć z dokładnością do trzeciego miejsca po przecinku 1 +/– 0,1 g (a gramów) próbki do szklanej rurki (5.3). Dodać 0,5 ml kwasu ortofosforowego (V) (4.4), 0,50 ml roztworu wewnętrznego standardu (4.6) i 9 ml alkoholu etylowego (4.1). Zamknąć rurkę i energicznie wytrząsnąć. Jeżeli to konieczne, umieścić rurkę w łaźni wodnej ogrzewanej do temperatury 60°C (5.4) na pięć minut w celu rozpuszczenia fazy lipidowej. Schłodzić szybko pod strumieniem bieżącej wody. Przesączyć część roztworu przez filtr membranowy (5.5). Zanalizować chromatograficznie przesącz tego samego dnia.
6.2. Warunki chromatografii gazowej
Poleca się następujące warunki operacyjne:
| Kolumna: |
|
|
|
| typ: | stal kwasoodporna |
|
| długość: | 2 m |
|
| średnica: | 1/8// |
|
| wypełnienie: | 10% SP™ 1000 (lub równoważne) + 1% H3PO4 na Chromosorbie WAW 100 do 120 mesh. |
| Temperatura: |
|
|
|
| dozownik | 200°C |
|
| kolumna | 120°C |
|
| detektor | 200°C |
| Gaz nośny: |
| azot |
| Szybkość przepływu: |
| 2,5 ml/min |
6.3. Analiza chromatograficzna.
6.3.1. Kalibrowanie
Przenieść pipetą odpowiednio 0,25; 0,50; 1,00; 2,00 i 4,00 ml roztworu kwasu propionowego (4.5) do serii 20 ml kolb miarowych. Do wszystkich kolb miarowych przenieść pipetą po 1,00 ml roztworu wewnętrznego standardu (4.6), uzupełnić objętość alkoholem etylowym (4.1) i wymieszać. Roztwory przygotowane w ten sposób zawierają „e” mg/ml kwasu 2-metylopropionowego jako wewnętrznego standardu (co oznacza 1 mg/ml, jeżeli „e” = 1000) i p/4; p/2; p; 2p; 4p mg/ml kwasu propionowego (co oznacza 0,25; 0,50; 1,00; 2,00; 4,00 mg/ml, jeżeli p = 1000). Wprowadzić po 1 µl wszystkich tych roztworów i sporządzić krzywą odwzorowania przez wykreślenie stosunku mas kwas propionowy / kwas metylopropionowy na osi odciętych i stosunek odpowiednich powierzchni pików na osi rzędnych. Trzykrotnie wprowadzić na kolumnę kolejno wszystkie roztwory i obliczyć średni stosunek pików.
6.3.2. Oznaczenie
Wprowadzić strzykawką 1 µl, przesączyć próbki (6.1.1). Porównać chromatogram z chromatogramem jednego z roztworów mianowanych (6.3.1). Jeżeli pik ma w przybliżeniu ten sam czas retencji jak kwas 2-metylopropionowy, należy zmienić wewnętrzny standard. Jeżeli nie obserwuje się żadnego nakładania się pików, wprowadzić 1 µl przesączu próbki (6.1.2) i zmierzyć powierzchnie piku kwasu propionowego i piku wewnętrznego standardu. Trzykrotnie wprowadzić na kolumnę kolejno wszystkie roztwory i obliczyć średni stosunek pików.
7. OBLICZENIA
7.1. Z krzywej kalibracyjnej otrzymanej w sposób opisany w pkt 6.3.1 wyznaczyć stosunek mas (K) odpowiadający stosunkowi powierzchni pików obliczonej w sposób opisany w pkt 6.3.2.
7.2. Z tak wyznaczonego stosunku mas obliczyć zawartość kwasu propionowego w próbce (x) jako procent masowy według wzoru:
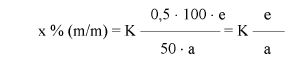
gdzie:
K = stosunek obliczony w sposób opisany w pkt 7.1,
e = masa w gramach wewnętrznego standardu odważona w sposób opisany w pkt 4.6,
a = masa w gramach próbki odważonej w sposób opisany w pkt 6.1.2.
Zaokrąglić wyniki do jednego miejsca dziesiętnego.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Dla zawartości kwasu propionowego 2% (m/m) różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać 0,12%.
XXXVII. IDENTYFIKOWANIE I OZNACZENIE HYDROCHINONU, ETERU MONOMETYLOWEGO HYDROCHINONU, ETERU MONOETYLOWEGO HYDROCHINONU I ETERU MONOBENZYLOWEGO HYDROCHINONU W PRODUKTACH KOSMETYCZNYCH
A. IDENTYFIKOWANIE
1. CEL I ZAKRES
Metoda opisuje wykrywanie i identyfikowanie hydrochinonu, eteru monometylowego hydrochinonu, eteru monoetylowego hydrochinonu i eteru monobenzylowego hydrochinonu.
2. ZASADA
Hydrochinon i jego etery identyfikuje się metodą chromatografii cienkowarstwowej (TLC).
3. ODCZYNNIKI
Wszystkie odczynniki muszą być czyste do analizy.
3.1. Alkohol etylowy, 96% (v/v).
3.2. Chloroform.
3.3. Eter dietylowy.
3.4. Rozpuszczalnik rozwijający: chloroform / eter dietylowy 66:33 (v/v).
3.5. Amoniak, 25% (m/m), d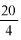
3.6. Kwas askorbinowy.
3.7. Hydrochinon.
3.8. Monometylowy eter hydrochinonu.
3.9. Monoetylowy eter hydrochinonu.
3.10. Monobenzylowy eter hydrochinonu.
3.11. Roztwory odniesienia.
Poniższe roztwory odniesienia powinny być świeżo przygotowane i są trwałe jeden dzień
3.11.1. Odważyć 0,05 g hydrochinonu (3.7) w 10 ml kalibrowanej probówce. Dodać 0,250 g kwasu askorbinowego (3.6) i 5 ml alkoholu etylowego (3.1). Dodawać amoniak (3.5) do uzyskania pH 10 i uzupełnić objętość do 10 ml alkoholem etylowym (3.1).
3.11.2. Odważyć 0,05 g eteru monometylowego hydrochinonu (3.9) w 10 ml kalibrowanej probówce. Dodać 0,250 g kwasu askorbinowego (3.6) i 5 ml alkoholu etylowego (3.1). Dodawać amoniak (3.5) do uzyskania pH 10 i uzupełnić objętość do 10 ml alkoholem etylowym (3.1).
3.11.3. Odważyć 0,05 g eteru monoetylowego hydrochinonu (3.8) w 10 ml kalibrowanej probówce. Dodać 0,250 g kwasu askorbinowego (3.6) i 5 ml alkoholu etylowego (3.1). Dodawać amoniak (3.5) do uzyskania pH 10 i uzupełnić objętość do 10 ml alkoholem etylowym (3.1).
3.11.4. Odważyć 0,05 g eteru monobenzylowego hydrochinonu (3.10) w 10 ml kalibrowanej probówce. Dodać 0,250 g kwasu askorbinowego (3.6) i 5 ml alkoholu etylowego (3.1). Dodawać amoniak (3.5) do uzyskania pH 10 i uzupełnić objętość do 10 ml alkoholem etylowym (3.1).
3.12. Azotan (V) srebra.
3.13. Kwas 12-molibdenofosforowy.
3.14. 6-hydrat heksacyjanożelazianu (III) potasu.
3.15. 6-hydrat chlorku żelaza (III).
3.16. Odczynniki wywołujące.
3.16.1. Do 5% (m/v) wodnego roztworu azotanu srebra (3.12) dodawać amoniak (3.5), aż powstający osad się rozpuści.
Ostrzeżenie: Roztwór staje się nietrwały i grozi wybuchem przy przechowywaniu i powinien być odrzucony po użyciu.
3.16.2. 10% (m/v) roztwór kwasu 12-molibdenofosforowego (3.13) w alkoholu etylowym (3.1).
3.16.3. Przygotować 1% (m/v) wodny roztwór heksacyjanożelazianu (III) potasu (3.14) i 2% (m/v) wodny roztwór chlorku żelaza (III) (3.15). Wymieszać równe części obu roztworów bezpośrednio przed użyciem.
4. APARATURA
Zwykły sprzęt laboratoryjny.
4.1. Typowy sprzęt do chromatografii cienkowarstwowej (TLC).
4.2. Płytki TLC, przygotowane fabrycznie: żel krzemionkowy GHR/UV254; 20 x 20 cm (Machery, Nagel lub równoważne). Grubość warstwy 0,25 mm.
4.3. Łaźnia ultradźwiękowa.
4.4. Wirówka.
4.5. Lampa UV, długość fali 254 nm.
5. PROCEDURA
5.1. Przygotowanie próbki
Odważyć 3,0 g próbki w 10 ml kalibrowanej rurce. Dodać 0,250 g kwasu askorbinowego (3.6) i 5 ml alkoholu etylowego (3.1). Doprowadzić odczyn roztworu do pH 10, dodając amoniak (3.5). Uzupełnić objętość do 10 ml alkoholem etylowym (3.1). Zamknąć rurkę korkiem i homogenizować roztwór w łaźni ultradźwiękowej w ciągu 10 minut. Przesączyć przez bibułę filtracyjną lub odwirować przy 3000 obr./min.
5.2. TLC
5.2.1. Wysycić komorę chromatograficzną rozpuszczalnikiem rozwijającym (3.4).
5.2.2. Nanieść na płytkę po 2 µl roztworów odniesienia (3.11) i 2 µl roztworu próbki (5.1). Chromatogram rozwijać w ciemności w temperaturze otoczenia aż do osiągnięcia przez czoło rozpuszczalnika wysokości 15 cm od linii początkowej.
5.2.3. Wyjąć płytkę i wysuszyć w temperaturze pokojowej.
5.3. Wykrywanie
5.3.1. Obserwować płytkę w świetle UV przy 254 nm i zaznaczyć położenie plam.
5.3.2. Spryskać płytkę:
1) odczynnikiem azotanu (V) srebra (3.16.1) lub
2) odczynnikiem kwasu 12-molibdenofosforowego (3.16.2); ogrzewać do temperatury ok. 120°C, lub
3) roztworem heksacyjanożelazianu (III) potasu i roztworu chlorku żelaza (III) (3.16.3).
6. IDENTYFIKOWANIE
Obliczyć wartość Rf dla wszystkich plam.
Porównać plamy otrzymane dla roztworu próbki z plamami otrzymanymi dla roztworów odniesienia pod względem ich wartości Rf kolorów plam w świetle UV i kolorów plam po ich wywołaniu odczynnikiem do spryskiwania. Wykonać analizę metodą HPLC opisaną w następnej części B i porównać czas retencji otrzymany dla piku (pików) próbki z czasem dla pików roztworów odniesienia. Wykorzystać wyniki z analizy metodą TLC i HPLC w celu zidentyfikowania obecności hydrochinonu i/lub jego eterów.
7. UWAGI
W opisanych warunkach otrzymano następujące wartości Rf:
| hydrochinon | 0,32 |
| eter monometylowy hydrochinonu | 0,53 |
| eter monoetylowy hydrochinonu | 0,55 |
| eter monobenzylowy hydrochinonu | 0,58 |
B. OZNACZANIE
1. CEL I ZAKRES
Przedstawiono szczegółowo procedurę oznaczania hydrochinonu, eteru monometylowego hydrochinonu, eteru monoetylowego hydrochinonu i eteru monobenzylowego hydrochinonu w produktach kosmetycznych do rozjaśniania skóry.
2. ZASADA
Próbkę ekstrahuje się mieszaniną woda/metanol podczas łagodnego ogrzewania w celu stopienia wszystkich ciekłych materiałów. Oznaczanie analizowanych substancji w powstałym roztworze wykonuje się metodą chromatografii cieczowej z zastosowaniem fazy odwróconej z detekcją w zakresie promieniowania UV.
3. ODCZYNNIKI
3.1. Wszystkie odczynniki powinny być jakości analitycznej. Stosowana woda musi być wodą destylowaną lub wodą o co najmniej równoważnej czystości.
3.2. Metanol.
3.3. Hydrochinon.
3.4. Eter monometylowy hydrochinonu.
3.5. Eter monoetylowy hydrochinonu.
3.6. Eter monobenzylowy hydrochinonu (monobenzon).
3.7. Tetrabydrofuran o jakości odpowiedniej dla HPLC.
3.8. Mieszanina woda/metanol 1:1 (v/v). Zmieszać jedną objętość wody z jedną objętością metanolu (3.2).
3.9. Faza ruchoma: mieszanina tetrahydrofuran/woda 45:55 (v/v). Zmieszać 45 objętości tetrahydrofuranu (3.7) i 55 objętości wody.
3.10. Roztwór odniesienia
Odważyć 0,06 g hydrochinonu (3.3), 0,08 g eteru monometylowego hydrochinonu (3.4), 0,10 g eteru monoetylowego hydrochinonu (3.5) i 0,12 g eteru monobenzylowego hydrochinonu (3.6) w 50 ml kolbie miarowej. Rozpuścić i uzupełnić objętość metanolem (3.2). Przygotować roztwór odniesienia przez rozcieńczenie 10,00 ml tego roztworu do 50 ml mieszaniną woda/metanol (3.8). Roztwory te muszą być świeżo przygotowane.
4. APARATURA
Zwykły sprzęt laboratoryjny.
4.1. Łaźnia wodna z możliwością utrzymania temperatury 60°C.
4.2. Wysokosprawny chromatograf cieczowy z detektorem UV o zmiennej długości fali i 10 µl pętlą do wprowadzania próbki.
4.3. Kolumna analityczna
Kolumna ze stali kwasoodpornej, długość 250 mm, średnica wewnętrzna 4,6 mm z wypełnieniem fenylowym Zorbax (chemicznie związany fenyloetylosilan osadzony na nośniku Zorbax SIL, na końcu osłonięty trimetylochlorosilanem), rozmiar cząsteczek 6 µm, lub równoważnym. Nie należy używać kolumny zabezpieczającej (ochronnej), z wyjątkiem ochrony fenylowej lub równoważnej.
4.4. Krążki bibuły filtracyjnej o średnicy 90 mm (Schleicher and Schüll, Weissband nr 5892 lub równoważna).
5. PROCEDURA
5.1. Przygotowanie próbki
Odważyć z dokładnością do trzeciego miejsca po przecinku 1 +/– 0,1 g (a gramów) próbki w 50 ml kolbie miarowej. Dyspergować próbkę w 25 ml mieszaniny woda/metanol (3.8). Zamknąć kolbę i wytrząsać energicznie do uzyskania jednorodnej zawiesiny. Wytrząsać w ciągu co najmniej jednej minuty. Umieścić kolbę w łaźni wodnej (4.1) utrzymywanej w temperaturze 60°C w celu poprawienia ekstrakcji. Schłodzić kolbę i uzupełnić objętość mieszaniną woda/metanol (3.8). Przesączyć ekstrakt, używając krążków bibuły filtracyjnej (4.4). Wykonać oznaczenie metodą HPLC w ciągu 24 godzin od przygotowania ekstraktu. 5.2. Wysokosprawna chromatografia cieczowa.
5.2.1. Uregulować szybkość przepływu fazy ruchomej (3.9) na 1,0 ml/min i nastawić długość fali detektora na 295 nm.
5.2.2. Wprowadzić strzykawką 10 µl roztworu próbki otrzymanego, jak opisano w pkt 5.1, i zarejestrować chromatogram. Zmierzyć powierzchnie pików. Przeprowadzić kalibrowanie według opisu w pkt 5.2.3. Porównać chromatogramy otrzymane dla roztworów próbki i roztworu mianowanego. Powierzchnie pików i współczynniki kalibracyjne RF obliczone zgodnie z pkt 5.2.3 zastosować do obliczenia stężeń analizowanych substancji w roztworze próbki.
5.2.3. Kalibrowanie
Wprowadzić strzykawką 10 µl roztworu odniesienia (3.10) i zarejestrować chromatogram.
Wprowadzać próbkę kilkakrotnie aż do otrzymania stałej powierzchni piku.
Oznaczyć współczynnik kalibracji RFi według wzoru:
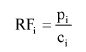
gdzie:
pi = powierzchnia piku dla hydrochinonu, eteru monometylowego hydrochinonu, eteru monoetylowego hydrochinonu lub eteru monobenzylowego hydrochinonu,
ci = stężenie (g / 50 ml) w roztworze odniesienia (3.10) hydrochinonu, eteru monometylowego hydrochinonu, eteru monoetylowego hydrochinonu lub eteru monobenzylowego hydrochinonu.
Należy upewnić się, czy chromatogramy otrzymane dla roztworu mianowanego i roztworu próbki odpowiadają następującym wymaganiom:
1) Rozdział pików w najgorzej rozdzielonej parze powinien wynosić co najmniej 0,90 (definicję rozdziału pików przedstawiono na rys. 15).
Rys. 15
Rozdział pików
Jeżeli nie uzyskano wymaganego rozdziału pików, należy zastosować kolumnę o większej rozdzielczości lub poprawiać skład fazy ruchomej, aż wymagania zostaną spełnione.
2) Współczynnik asymetrii As wszystkich otrzymanych pików powinien znajdować się w zakresie od 0,9 do 1,5 (definicję współczynnika asymetrii pików przedstawiono na rys. 16). Do zarejestrowania chromatogramu w celu oznaczenia współczynnika asymetrii jest polecana szybkość przesuwu papieru co najmniej 2 cm/min.
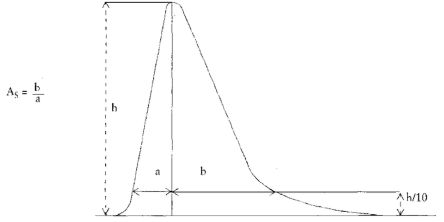
Rys. 16
Współczynnik asymetrii piku
3) Otrzymuje się stałą linię podstawową.
6. OBLICZENIE
Powierzchnie pików analizowanych substancji stosuje się do obliczenia stężenia (stężeń) tej (tych) substancji w próbce. Obliczyć stężenie analizowanej substancji w próbce w procentach masowych (xi) według wzoru:
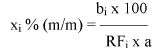
gdzie:
a = masa analizowanej próbki w gramach,
bi = powierzchnia piku substancji analizowanej „i” w próbce.
7. POWTARZALNOŚĆ (patrz: norma ISO 5725)
7.1. Dla zawartości hydrochinonu 2,0% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,13%.
7.2. Dla zawartości eteru monometylowego hydrochinonu 1,0% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,1%.
7.3. Dla zawartości eteru monoetylowego hydrochinonu 1,0% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,11%.
7.4. Dla zawartości eteru monobenzylowego hydrochinonu 1,0% różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie powinna przekraczać absolutnej wartości 0,11%.
8. ODTWARZALNOŚĆ (patrz: norma ISO 5725)
8.1. Dla zawartości hydrochinonu 2,0% różnica między wynikami dwóch oznaczeń wykonanych dla tej samej próbki w różnych warunkach (inne laboratoria, inni wykonawcy, inna aparatura i/lub inny czas) nie powinna przekraczać absolutnej wartości 0,37%.
8.2. Dla zawartości eteru monometylowego hydrochinonu 1,0% różnica między wynikami dwóch oznaczeń wykonanych dla tej samej próbki w różnych warunkach (inne laboratoria, inni wykonawcy, inna aparatura i/lub inny czas) nie powinna przekraczać absolutnej wartości 0,21%.
8.3. Dla zawartości eteru monoetylowego hydrochinonu 1,0% różnica między wynikami dwóch oznaczeń wykonanych dla tej samej próbki w różnych warunkach (inne laboratoria, inni wykonawcy, inna aparatura i/lub inny czas) nie powinna przekraczać absolutnej wartości 0,19%.
8.4. Dla zawartości eteru monobenzylowego hydrochinonu 1,0% różnica między wynikami dwóch oznaczeń wykonanych dla tej samej próbki w różnych warunkach (inne laboratoria, inni wykonawcy, inna aparatura i/lub inny czas) nie powinna przekraczać absolutnej wartości 0,11%.
9. UWAGI
9.1. Jeżeli stwierdzono zawartość hydrochinonu znacznie ponad 2%, a jest wymagana dokładna ocena zawartości, ekstrakt próbki (5.1) powinno się rozcieńczyć do stężenia podobnego do tego, jakie można by otrzymać z próbki zawierającej 2% hydrochinonu, i powtórzyć oznaczenie (dla wysokich stężeń hydrochinonu, w niektórych przyrządach absorbancja może znaleźć się poza liniowym zakresem wskazań detektora).
9.2. Zakłócenia
Opisana powyżej metoda pozwala na oznaczenie hydrochinonu i jego eterów w jednym izokratycznym przebiegu. Zastosowanie kolumny fenylowej zapewnia wystarczającą zdolność zatrzymywania hydrochinonu, czego nie można zagwarantować przy zastosowaniu kolumny Cl8 z użyciem podanej fazy ruchomej. Jednakże metoda ta jest podatna na zakłócenia przez szereg parabenów (estrów kwasu 4-hydroksybenzoesowego). W takich przypadkach należy powtórzyć oznaczenie z różnymi układami faza ruchoma / faza stacjonarna.
Kolumna: Zorbax ODS, 4,6 mm x 25 mm lub równoważna:
temperatura: 36°C
szybkość przepływu: 1,5 ml/min
faza ruchoma:
dla hydrochinonu: metanol/woda 5/95 (v/v)
dla eteru monometylowego hydrochinonu: metanol/woda 30/70 (v/v)
dla eteru monobenzylowego hydrochinonu: metanol/woda 80/20 (v/v)
Kolumna: Spherisorb S5-ODS lub równoważna:
faza ruchoma: woda/metanol 90/10 (v/v)
szybkość przepływu: 1,5 ml/min
Te warunki są odpowiednie dla hydrochinonu.
XXXVIII. IDENTYFIKOWANIE I OZNACZANIE 2-FENOKSYETANOLU, 1-FENOKSYPROPAN-2-OLU, 4-HYDROKSYBENZOESANÓW METYLU, ETYLU, PROPYLU, BUTYLU I BENZYLU W PRODUKTACH KOSMETYCZNYCH
A. IDENTYFIKOWANIE
1. PRZEDMIOT I ZAKRES STOSOWANIA
W metodzie określa się procedurę TLC, która w połączeniu z metodą oznaczania opisaną w sekcji B, pozwala na identyfikowanie 2-fenoksyetanolu, 1-fenoksypropan-2-olu, 4-hydroksybenzoesanu metylu, 4-hydroksybenzoesanu etylu, 4-hydroksybenzoesanu propylu, 4-hydroksybenzoesanu butylu i 4-hydroksybenzoesanu benzylu w produktach kosmetycznych.
2. ZASADA
Środki konserwujące ekstrahuje się acetonem z zakwaszonej próbki produktu kosmetycznego. Po przesączeniu roztwór acetonowy miesza się z wodą i w środowisku alkalicznym kwasy tłuszczowe zostają strącone w postaci soli wapnia. Alkaliczną mieszaninę aceton/woda ekstrahuje się eterem dietylowym w celu usunięcia substancji lipofilowych. Po zakwaszeniu środki konserwujące ekstrahuje się eterem dietylowym. Próbkę ekstraktu w eterze dietylowym nanosi się na płytkę do chromatografii cienkowarstwowej pokrytą żelem krzemionkowym. Po rozwinięciu chromatogram otrzymany na płytce obserwuje się w świetle UV i wywołuje, używając odczynnika Millona.
3. ODCZYNNIKI
3.1. Ogólne
Wszystkie używane odczynniki posiadają czystość analityczną. Woda jest wodą destylowaną lub wodą co najmniej równej czystości.
3.2. Aceton.
3.3. Eter dietylowy.
3.4. n-Pentan.
3.5. Metanol.
3.6. Kwas octowy, lodowaty.
3.7. Kwas solny, roztwór c(HCl) = 4 mol/l.
3.8. Wodorotlenek potasu, roztwór c(KOH) = 4 mol/l.
3.9. 2-hydrat chlorku wapnia (CaCl2 2H2O).
3.10. Odczynnik wywołujący: odczynnik Millona
Odczynnik Millona [azotan rtęci (II)] to fabrycznie przygotowany roztwór, dostępny w handlu (Fluka 69820).
3.11. 2-Fenoksyetanol.
3.12. 1-Fenoksypropan-2-ol.
3.13. 4-Hydroksybenzoesan metylu (metyloparaben).
3.14. 4-Hydroksybenzoesan etylu (etyloparaben).
3.15. 4-Hydroksybenzoesan n-propylu (propyloparaben).
3.16. 4-Hydroksybenzoesan n-butylu (butyloparaben).
3.17. 4-Hydroksybenzoesan benzylu (benzyloparaben).
3.18. Roztwory odniesienia
Przygotować 0,1% (m/V) roztwory wszystkich substancji odniesienia wymienionych w pkt 3.11, 3.12, 3.13, 3.14, 3.15, 3.16 i 3.17 w metanolu.
3.19. Rozpuszczalnik rozwijający
Zmieszać 88 objętości n-pentanu (3.4) z 12 objętościami kwasu octowego lodowatego (3.6).
4. APARATURA
Standardowe wyposażenie laboratoryjne.
4.1. Łaźnia wodna, z możliwością utrzymania temperatury 60°C.
4.2. Komora chromatograficzna (niewyłożona bibułą filtracyjną).
4.3. Źródło światła ultrafioletowego, 254 nm.
4.4. Płytki do chromatografii cienkowarstwowej, 20 cm x 20 cm, pokryte fabrycznie 0,25 mm warstwą żelu krzemionkowego 60 F254 ze strefą nanoszenia (Merck nr 11798 lub równoważne).
4.5. Suszarka z możliwością utrzymania temperatury 105°C.
4.6. Suszarka do suszenia włosów gorącym powietrzem.
4.7. Wełniany wałek do malowania, długość ok. 10 cm, średnica zewnętrzna ok. 3,5 cm. Grubość wełnianej warstwy powinna wynosić 2–3 mm. Jeżeli konieczne, należy przyciąć wełnianą warstwę (patrz: uwaga w pkt 5.2).
4.8. 50 ml szklana rurka z gwintowanym korkiem.
4.9. Płytka ogrzewana elektrycznie z termostatyczną kontrolą temperatury
Nastawić temperaturę ok. 80°C. Płytka grzejna jest pokryta płytką aluminiową o wymiarach 20 cm x 20 cm i o grubości 6 mm dla uzyskania równomiernego rozprowadzania ciepła.
5. PROCEDURA
5.1. Przygotowanie próbki
Odważyć ok. 1 g próbki w 50 ml szklanej rurce z gwintowanym korkiem (4.8). Dodać cztery krople roztworu kwasu solnego (3.7) i 40 ml acetonu.
Do produktów kosmetycznych silnie alkalicznych, takich jak mydło toaletowe, dodaje się 20 kropli roztworu kwasu solnego. Zamknąć rurkę, łagodnie ogrzewać mieszaninę do temperatury ok. 60°C dla ułatwienia ekstrakcji środków konserwujących do fazy acetonowej i wytrząsać energicznie przez jedną minutę.
Zmierzyć pH roztworu papierkiem wskaźnikowym pH i dostosować kwas solny do pH ≤ 3. Wytrząsać energicznie ponownie przez jedną minutę.
Schłodzić roztwór do temperatury pokojowej i przesączyć przez bibułę filtracyjną do kolby stożkowej. Przenieść 20 ml przesączu do 200 ml kolby stożkowej, dodać 60 ml wody i wymieszać. Doprowadzić odczyn mieszaniny do pH 10, dodając wodorotlenek potasu (3.8), używając papierka wskaźnikowego pH.
Dodać 1 g 2-hydratu chlorku wapnia (3.9) i wytrząsać energicznie. Przesączyć roztwór przez bibułę filtracyjną do 250 ml lejka rozdzielczego zawierającego 75 ml eteru dietylowego i wytrząsać energicznie przez jedną minutę.
Pozostawić warstwy do rozdzielenia i zebrać warstwę wodną w 200 ml kolbie stożkowej. Doprowadzić odczyn roztworu do pH ok. 2, dodając roztwór kwasu solnego, używając papierka wskaźnikowego pH. Następnie dodać 10 ml eteru dietylowego i wytrząsać energicznie przez jedną minutę. Pozostawić do rozdzielenia warstw i przenieść ok. 2 ml warstwy eteru dietylowego do 5 ml fiolki na próbkę.
5.2. Chromatografia cienkowarstwowa (TLC).
Umieścić płytkę do chromatografii cienkowarstwowej (4.4) na ogrzewanej płytce aluminiowej (4.9). Nanieść po 10 µl kolejno wszystkich roztworów odniesienia (3.18) i 100 µl roztworu (roztworów) próbki (próbek) (5.1) na linię początkową w strefie nanoszenia na płytce do chromatografii cienkowarstwowej.
Jeżeli jest to pożądane, można użyć strumienia powietrza dla ułatwienia odparowania rozpuszczalnika. Zdjąć płytkę TLC z ogrzewanej płytki i pozostawić do schłodzenia do temperatury pokojowej. Przenieść 100 ml rozpuszczalnika rozwijającego (3.19) do komory chromatograficznej (4.2).
Niezwłocznie umieścić płytkę TLC w nienasyconej komorze i rozwijać w temperaturze pokojowej do chwili, gdy czoło rozpuszczalnika osiągnie odległość ok. 15 cm od linii bazowej.
Wyjąć płytkę z komory chromatograficznej i wysuszyć w strumieniu gorącego powietrza za pomocą suszarki do włosów z gorącym powietrzem.
Zbadać płytkę w świetle UV (4.3) i zaznaczyć położenie plam. Ogrzewać płytkę w ciągu 30 minut w suszarce (4.5) w temperaturze 100°C dla usunięcia nadmiaru kwasu octowego. Wywołać plamy środków konserwujących na chromatogramie odczynnikiem Millona (3.10) przez zanurzenie wałka do malowania (4.7) w odczynniku i przesunięcie po płytce TLC do równomiernego nawilżenia.
Uwaga: Alternatywnie plamy można wywołać przez staranne spryskanie odczynnikiem Millona wszystkich plam zaznaczonych w świetle UV.
Estry kwasu 4-hydroksybenzoesowego pojawiają się jako czerwone plamy, 2-fenoksyetanol i 1-fenoksypropan-2-ol jako żółte plamy. Jednak należy zauważyć, że sam kwas 4-hydroksybenzoesowy, który może znajdować się w próbkach jako środek konserwujący lub produkt rozkładu parabenu, pojawi się również jako czerwona plama (7.3 i 7.4).
6. IDENTYFIKOWANIE
Obliczyć wartości Rf dla wszystkich plam. Porównać plamy otrzymane z roztworu próbki z plamami roztworów odniesienia ze względu na ich wartości Rf, zachowanie w promieniowaniu UV i kolor po wywołaniu. Wyciągnąć wstępne wnioski dotyczące identyfikowania środków konserwujących. Jeżeli zachodzi przypuszczenie, że parabeny znajdują się w próbce, należy zastosować chromatografię cieczową HPLC opisaną w sekcji B. Połączyć wyniki otrzymane z TLC i wysokosprawną chromatografią cieczową (HPLC) dla potwierdzenia obecności 2-fenoksyetanolu, 1-fenoksypropan-2-olu i parabenów.
7. UWAGI
7.1. Ze względu na toksyczne działanie odczynnika Millona najlepszym sposobem jego stosowania jest jedna z opisanych procedur. Spryskiwanie nie jest polecane.
7.2. Inne związki zawierające grupy hydroksylowe mogą również dawać barwne plamy z odczynnikiem Millona.
7.3. Wartości Rf podane w tabeli poniżej służą jako wskazówka do identyfikacji plam.
| Związek | Rf x 100 | Barwa |
| kwas 4-hydroksybenzoesowy | 11 | czerwona |
| metyloparaben | 12 | czerwona |
| etyloparaben | 17 | czerwona |
| propyloparaben | 21 | czerwona |
| butyloparaben | 26 | czerwona |
| benzyloparaben | 16 | czerwona |
| 2-fenoksyetanol | 29 | żółta |
| 1-fenoksypropan-2-ol | 50 | żółta |
7.4. Nie uzyskuje się rozdziału kwasu 4-hydroksybenzoesowego i metyloparabenu lub benzyloparabenu i etyloparabenu. Identyfikowanie tych związków powinno być potwierdzone przez przeprowadzenie analizy metodą HPLC opisaną w sekcji B i porównanie czasów retencji otrzymanych dla próbki z czasami dla norm.
B. OZNACZANIE
1. PRZEDMIOT I ZAKRES ZASTOSOWANIA
Metodę tę stosuje się do oznaczania 2-fenoksyetanolu, 1-fenoksypropan-2-olu, 4-hydroksybenzoesanu metylu, 4-hydroksybenzoesanu etylu, 4-hydroksybenzoesanu propylu, 4-hydroksybenzoesanu butylu i 4-hydroksybenzoesanu benzylu w produktach kosmetycznych.
2. DEFINICJE
Ilości środków konserwujących oznaczone tą metodą są wyrażone w procentach masowych.
3. ZASADA
Próbkę zakwasza się, dodając kwas siarkowy i następnie dysperguje w mieszaninie alkoholu etylowego i wody. Po łagodnym podgrzaniu mieszaninę filtruje się w celu stopienia fazy ciekłej dla poprawienia ilościowej ekstrakcji.
Środki konserwujące w przesączu oznacza się metodą wysokosprawnej chromatografii cieczowej (HPLC) na odwróconych fazach, stosując 4-hydroksybenzoesan izopropylu jako standard wewnętrzny.
4. ODCZYNNIKI
4.1. Ogólne
Wszystkie odczynniki muszą mieć czystość analityczną i być – tam, gdzie to stosowne – odpowiednie do HPLC. Woda powinna być wodą destylowaną lub wodą o co najmniej równej czystości.
4.2. Alkohol etylowy, absolutny.
4.3. 2-Fenoksyetanol.
4.4. 1-Fenoksybenzoesan-2-ol.
4.5. 4-Hydroksybenzoesan metylu (metyloparaben).
4.6. 4-Hydroksybenzoesan etylu (etyloparaben).
4.7. 4-Hydroksybenzoesan n-propylu (propyloparaben).
4.8. 4-Hydroksybenzoesan izopropylu (izopropyloparaben).
4.9. 4-Hydroksybenzoesan n-butylu (butyloparaben).
4.10. 4-Hydroksybenzoesan benzylu (benzyloparaben).
4.11. Tetrahydrofuran.
4.12. Metanol.
4.13. Acetonitryl.
4.14. Kwas siarkowy, roztwór c (H2SO4) = 2 mole/l.
4.15. Mieszanina alkohol etylowy/woda
Zmieszać dziewięć objętości alkoholu etylowego (4.2) i jedną objętość wody.
4.16. Roztwór wewnętrznego standardu
Odważyć dokładnie ok. 0,25 g izopropyloparabenu (4.8), przenieść do 500 ml kolby miarowej, rozpuścić i uzupełnić objętość mieszaniną alkohol etylowy/woda (4.15).
4.17. Faza ruchoma: mieszanina tetrahydrofuran/woda/metanol/acetonitryl
Zmieszać 5 objętości tetrahydrofuranu, 60 objętości wody, 10 objętości metanolu i 25 objętości acetonitrylu.
4.18. Bazowy roztwór środków konserwujących
Do 100 ml kolby miarowej odważyć dokładnie ok. 0,2 g 2-fenoksyetanolu, 0,2 g 1-fenoksypropan-2-olu, 0,05 g metyloparabenu, 0,05 g etyloparabenu, 0,05 g propyloparabenu, 0,05 g butyloparabenu i 0,025 g benzyloparabenu, rozpuścić i uzupełnić objętość mieszaniną etanol/woda. Roztwór przechowywany w lodówce jest trwały przez jeden tydzień.
4.19. Standardowe roztwory środków konserwujących.
Z roztworu bazowego (4.18) przenieść odpowiednio 20,00 ml, 10,00 ml, 5,00 ml, 2,00 ml i 1,00 ml do 50 ml kolb miarowych. Do każdej kolby dodać po 10,00 ml roztworu wewnętrznego standardu (4.16) i 1,0 ml roztworu kwasu siarkowego (4.14) i uzupełnić objętość mieszaniną alkohol etylowy/woda. Roztwory te powinny być świeżo przygotowane.
5. APARATURA
Standardowe wyposażenie laboratoryjne.
5.1. Łaźnia wodna, z możliwością utrzymania temperatury 60°C +/– 1°C.
5.2. Wysokosprawny chromatograf cieczowy z detektorem UV o długości fali 280 nm.
5.3. Kolumna analityczna
Stal kwasoodporna, długość 25 cm, średnica wewnętrzna 4,6 mm (lub długość 12,5 cm, średnica wewnętrzna 4,6 mm) z wypełnieniem Nucleosil 5C18 lub równoważnym (10.1).
5.4. 100 ml szklane rurki z gwintowanym korkiem.
5.5. Kamyki wrzenne, z karborundu, o wymiarach 2 do 4 mm lub równoważne.
6. PROCEDURA
6.1. Przygotowanie próbki
6.1.1. Przygotowanie próbki bez dodatku wewnętrznego standardu.
Do 100 ml szklanej rurki z gwintowanym korkiem odważyć ok. 1,0 g próbki. Pipetą dodać do rurki 1,0 ml roztworu kwasu siarkowego (4.14) i 50 ml mieszaniny alkohol etylowy/woda (4.15). Dodać ok. 1 g kamyków wrzennych (5.5), zamknąć rurkę i wytrząsać energicznie aż do otrzymania homogenicznej zawiesiny. Wytrząsać co najmniej przez jedną minutę. Umieścić rurkę na pięć minut w łaźni wodnej (5.1) utrzymywanej w temperaturze 60°C +/– 1°C dla ułatwienia ekstrakcji środków konserwujących do fazy etanolowej.
Niezwłocznie schłodzić rurkę w strumieniu zimnej wody i przechowywać ekstrakt w lodówce przez jedną godzinę. Przesączyć ekstrakt przez bibułę filtracyjną. Przenieść ok. 2 ml przesączu do 5 ml fiolki na próbkę. Ekstrakty należy przechowywać w lodówce i oznaczyć skład metodą HPLC w ciągu 24 godzin.
6.1.2. Przygotowanie próbki z dodatkiem wewnętrznego standardu.
Do 100 ml szklanej rurki z gwintowanym korkiem odważyć z dokładnością do trzeciego miejsca dziesiętnego 1,0 +/– 0,1 g próbki.
Do rurki dodać za pomocą pipety 1,0 ml roztworu kwasu siarkowego i 40,0 ml mieszaniny alkohol etylowy/woda. Dodać ok. 1 g kamyków wrzennych (5.5) i dokładnie 10,00 ml roztworu wewnętrznego standardu. Zamknąć rurkę i wstrząsać energicznie aż do otrzymania homogenicznej zawiesiny. Wstrząsać przez co najmniej jedną minutę. Umieścić rurkę na okres pięciu minut w łaźni wodnej utrzymywanej w temperaturze 60°C +/– 1°C w celu ułatwienia ekstrakcji środków konserwujących do fazy etanolowej.
Niezwłocznie schłodzić rurkę w strumieniu bieżącej zimnej wody i przechowywać ekstrakt w lodówce przez jedną godzinę. Przesączyć ekstrakt przez bibułę filtracyjną. Przenieść ok. 2 ml przesączu do 5 ml fiolki na próbę (roztwór testowy). Przechowywać ekstrakt w lodówce i wykonać oznaczenia metodą HPLC w ciągu 24 godzin.
6.2. Wysokosprawna chromatografia cieczowa (HPLC).
6.2.1. Warunki analizy chromatograficznej.
Faza ruchoma: mieszanina tetrahydrofuran/woda/metanol/acetonitryl (4.17).
Szybkość przepływu: 1,5 ml/minutę.
Długość fali detekcji: 280 mm.
6.2.2. Kalibrowanie
Wstrzyknąć po 10 µl wszystkich standardowych roztworów środków konserwujących (4.19). Z otrzymanych chromatogramów oznaczyć stosunki wysokości szczytu standardowych roztworów środków konserwujących do wysokości szczytu wewnętrznego standardu. Wykreślić krzywą dla każdego środka konserwującego odnoszącą te stosunki do stężeń roztworów standardowych.
6.2.3. Oznaczanie
Wstrzyknąć po 10 µl roztworu próbki bez wewnętrznego standardu (6.1.1) do chromatografu i zarejestrować chromatogram.
Wstrzyknąć 10 µl jednego ze standardowych roztworów środków konserwujących (4.19) i zarejestrować chromatogram. Porównać otrzymane chromatogramy.
Jeżeli na chromatogramie ekstraktu próbki (6.1.1) nie ma żadnego szczytu mającego w przybliżeniu ten sam czas retencji jak izopropyloparaben (polecany standard wewnętrzny), kontynuować, wprowadzając strzykawką 10 µl roztworu próbki z wewnętrznym standardem (6.1.2). Zarejestrować chromatogram i zmierzyć wysokości szczytu.
Jeżeli na chromatogramie roztworu próbki występuje szczyt przeszkadzający o takim samym czasie retencji jak izopropyloparaben, należy wybrać inny wewnętrzny standard.
Jeżeli jeden ze środków konserwujących poddawanych badaniu jest nieobecny na chromatogramie, ten środek można stosować jako alternatywny wewnętrzny standard. Obliczyć stosunki wysokości szczytów badanych środków konserwujących do wysokości szczytu wewnętrznego standardu.
Upewnić się, że dla roztworów standardowych stosowanych w procedurze kalibrowania otrzymuje się liniową zależność.
Upewnić się, że chromatogramy otrzymane dla roztworu standardowego i roztworu próbki odpowiadają następującym wymaganiom:
1) Rozdział szczytów najgorzej rozdzielonej pary powinien wynosić co najmniej 0,90 (definicję rozdziału pików przedstawiono na rys. 17).
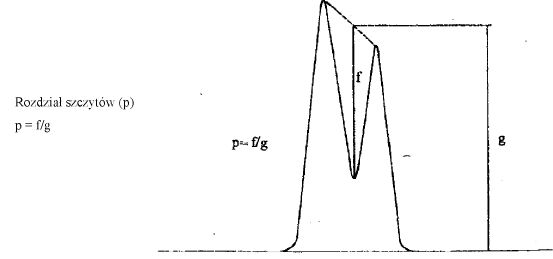
Rys. 17
Rozdział szczytów
Jeżeli nie uzyskano wystarczającego rozdziału, można zastosować kolumnę o większej rozdzielczości lub poprawiać skład fazy ruchomej, aż zostaną spełnione wymagania.
2) Współczynniki asymetrii As wszystkich otrzymanych szczytów znajdują się w zakresie 0,9–1,5 (definicję współczynnika asymetrii przedstawiono na rys. 18) Do zarejestrowania chromatogramu w celu oznaczenia współczynnika asymetrii zaleca się szybkość przesuwu papieru co najmniej 2 cm/min.
Współczynnik asymetrii szczytu As
As = b/a
Rys. 18
Współczynnik asymetrii szczytu As
3) Powinno się otrzymać stałą linię podstawową.
7. OBLICZENIE
Zastosować krzywą odwzorowania (6.2.2) i stosunki wysokości szczytów badanych środków konserwujących do wysokości szczytu wewnętrznego standardu do obliczenia stężenia środków konserwujących w roztworze próbki. Obliczyć zawartość 2-fenoksyetanolu, 1-fenoksypropan-2-olu, 4-hydroksybenzoesanu metylu, 4-hydroksybenzoesanu etylu, 4-hydroksybenzoesanu propylu, 4-hydroksybenzoesanu butylu i 4-hydroksybenzoesanu benzylu, wi, w procentach masowych (% m/m) według wzoru:
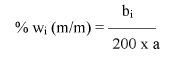
gdzie:
bi = stężenie (µg/ml) środka konserwującego „i” w roztworze badanym odczytane z krzywej odwzorowania,
a = masa (g) analizowanej próbki.
8. POWTARZALNOŚĆ (patrz: norma ISO 5725)
Patrz: uwagi w pkt 10.5.
9. ODTWARZALNOŚĆ (patrz: norma ISO 5725)
Patrz: uwagi w pkt 10.5.
10. UWAGI
10.1. Faza spoczynkowa
Zachowanie retencyjne roztworu przy oznaczaniu metodą HPLC zależy w dużym stopniu od rodzaju, gatunku i przygotowania fazy spoczynkowej. O tym, czy kolumnę można zastosować do rozdziału badanych środków konserwujących, można wnioskować na podstawie wyników otrzymanych dla roztworów standardowych (patrz: uwagi w pkt 6.2.3). Stwierdzono, że poza proponowanymi już materiałami do wypełnienia kolumn również Hypersil ODS i Zorbax ODS są odpowiednie. Także skład polecanej fazy ruchomej można optymalizować w celu uzyskania wymaganego rozdziału.
10.2. Długość fali detekcji
Badanie chropowatości opisanej metody wykazało, że niewielka zmiana w długości fali detekcji może mieć znaczący wpływ na wyniki oznaczenia. Dlatego parametr ten musi być starannie kontrolowany podczas analizy.
10.3. Zakłócenia
W warunkach opisanych w tej metodzie wiele innych różnych związków, takich jak środki konserwujące i dodatki do produktów kosmetycznych, ulega równie dobrze eluowaniu.
10.4. Do zamontowanej kolumny analitycznej można użyć odpowiedniej kolumny zabezpieczającej.
10.5. Metodę sprawdzono we wspólnym doświadczeniu z udziałem dziewięciu laboratoriów. Analizowano trzy próbki. Poniższa tabela podaje zawartość w % m/m (m), powtarzalność (r) i odtwarzalność (R) substancji obecnych we wszystkich trzech próbkach.
| Próbka | 2-fenoksyetanol | 1-fenoksypropan-2-ol | metyloparaben | etyloparaben | propylparaben | butyloparaben | benzyloparaben | |
| Krem witaminowy | m | 1,124 |
| 0,250 | 0,0628 | 0,0310 | 0,0906 |
|
| r | 0,016 |
| 0,018 | 0,0035 | 0,0028 | 0,0044 |
| |
| R | 0,176 |
| 0,030 | 0,0068 | 0,0111 | 0,0034 |
| |
| Krem nawilżający | m | 1,196 |
| 0,266 | 0,076 |
|
|
|
| r | 0,040 |
| 0,003 | 0,002 |
|
|
| |
| R | 0,147 |
| 0,022 | 0,004 |
|
|
| |
| Krem do masażu | m |
| 0,806 |
|
| 0,180 | 0,148 | 0,152 |
| r |
| 0,067 |
|
| 0,034 | 0,013 | 0,015 | |
| R |
| 0,112 |
|
| 0,078 | 0,012 | 0,016 | |
1) Ze względu na bogatą gamę produktów kosmetycznych, w których może znajdować się heksachlorofen, ważną rzeczą jest sprawdzenie odzyskiwania heksachlorofenu z próbki według powyższej procedury przed zarejestrowaniem wyników. Jeżeli odzyskiwane są niewielkie ilości, to modyfikacje takie jak zmiana rozpuszczalnika (benzen zamiast octanu etylu) itp. mogłyby być wprowadzone za zgodą zainteresowanych stron.
2) Trwałość żółtego koloru wskazuje na nadmiar diazometanu, który jest niezbędny do zapewnienia całkowitego zmetylowania próbki.
- Data ogłoszenia: 2020-05-26
- Data wejścia w życie: 2020-05-27
- Data obowiązywania: 2020-05-27

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA