REKLAMA
Dziennik Ustaw - rok 2016 poz. 1536
OBWIESZCZENIE
MARSZAŁKA SEJMU RZECZYPOSPOLITEJ POLSKIEJ
z dnia 23 sierpnia 2016 r.
w sprawie ogłoszenia jednolitego tekstu ustawy o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych
1. Na podstawie art. 16 ust. 1 zdanie pierwsze ustawy z dnia 20 lipca 2000 r. o ogłaszaniu aktów normatywnych i niektórych innych aktów prawnych (Dz. U. z 2016 r. poz. 296) ogłasza się w załączniku do niniejszego obwieszczenia jednolity tekst ustawy z dnia 12 maja 2011 r. o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych (Dz. U. z 2015 r. poz. 345), z uwzględnieniem zmian wprowadzonych:
1) ustawą z dnia 9 października 2015 r. o zmianie ustawy o terminach zapłaty w transakcjach handlowych, ustawy – Kodeks cywilny oraz niektórych innych ustaw (Dz. U. poz. 1830),
2) ustawą z dnia 9 października 2015 r. o zmianie ustawy o systemie informacji w ochronie zdrowia oraz niektórych innych ustaw (Dz. U. poz. 1991 oraz z 2016 r. poz. 580 i 832),
3) ustawą z dnia 18 marca 2016 r. o zmianie ustawy o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych oraz niektórych innych ustaw (Dz. U. poz. 652)
oraz zmian wynikających z przepisów ogłoszonych przed dniem 1 sierpnia 2016 r.
2. Podany w załączniku do niniejszego obwieszczenia jednolity tekst ustawy nie obejmuje:
1) art. 56 i art. 57 ustawy z dnia 9 października 2015 r. o zmianie ustawy o terminach zapłaty w transakcjach handlowych, ustawy – Kodeks cywilny oraz niektórych innych ustaw (Dz. U. poz. 1830), które stanowią:
„Art. 56. Do odsetek należnych za okres kończący się przed dniem wejścia w życie niniejszej ustawy stosuje się przepisy dotychczasowe.
Art. 57. Ustawa wchodzi w życie z dniem 1 stycznia 2016 r., z wyjątkiem art. 50, art. 51 i art. 54, które wchodzą w życie z dniem następującym po dniu ogłoszenia.”;
2) art. 18, art. 45, art. 48 ust. 1, art. 50 i art. 52 ustawy z dnia 9 października 2015 r. o zmianie ustawy o systemie informacji w ochronie zdrowia oraz niektórych innych ustaw (Dz. U. poz. 1991 oraz z 2016 r. poz. 580 i 832), które stanowią:
„Art. 18. Do dnia 31 grudnia 2017 r. wnioski, załączniki i dokumenty, o których mowa w art. 24 ust. 6 ustawy, o której mowa w art. 13, w brzmieniu nadanym niniejszą ustawą, mogą być składane w dotychczasowy sposób albo za pomocą Systemu Obsługi List Refundacyjnych.”
„Art. 45. 1. Umowy, o których mowa w art. 48 ustawy, o której mowa w art. 13, wygasają z dniem 31 grudnia 2016 r.
2. Do kontroli umów, o których mowa w ust. 1, stosuje się przepisy dotychczasowe.”
Art. 48. „1. Akty wykonawcze wydane na podstawie:
1) art. 18 ustawy, o której mowa w art. 1, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 18 ustawy, o której mowa w art. 1, w brzmieniu nadanym niniejszą ustawą,
2) art. 22 ust. 7 ustawy, o której mowa w art. 1, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 22 ust. 7 ustawy, o której mowa w art. 1, w brzmieniu nadanym niniejszą ustawą,
3) art. 28 ust. 3 ustawy, o której mowa w art. 1, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 28 ust. 3 ustawy, o której mowa w art. 1, w brzmieniu nadanym niniejszą ustawą,
4) art. 31 ustawy, o której mowa w art. 1, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 31 ustawy, o której mowa w art. 1, w brzmieniu nadanym niniejszą ustawą,
4a) art. 14e ust. 1 ustawy, o której mowa w art. 4, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 14f ust. 1 ustawy, o której mowa w art. 4, w brzmieniu nadanym niniejszą ustawą,
4b) art. 16 ust. 10, art. 16g ust. 1 i art. 16x ust. 1 ustawy, o której mowa w art. 4, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 16 ust. 10, art. 16g ust. 1 i art. 16x ust. 1 ustawy, o której mowa w art. 4, w brzmieniu nadanym niniejszą ustawą,
4c) art. 16r ust. 13 ustawy, o której mowa w art. 4, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 16ra ust. 2 ustawy, o której mowa w art. 4, w brzmieniu nadanym niniejszą ustawą,
4d) art. 16t ust. 4 ustawy, o której mowa w art. 4, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 16x ust. 1 ustawy, o której mowa w art. 4, w brzmieniu nadanym niniejszą ustawą,
5) art. 45 ust. 5 ustawy, o której mowa w art. 4, oraz art. 15a ust. 8 pkt 3–6 ustawy, o której mowa w art. 15, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 96a ust. 12 ustawy, o której mowa w art. 6, w brzmieniu nadanym niniejszą ustawą,
6) art. 28 ust. 3 ustawy, o której mowa w art. 6, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 28 ust. 3 ustawy, o której mowa w art. 6, w brzmieniu nadanym niniejszą ustawą,
7) art. 96 ust. 7 ustawy, o której mowa w art. 6, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 96 ust. 9 i 10 ustawy, o której mowa w art. 6, w brzmieniu nadanym niniejszą ustawą,
8) art. 38 ust. 7 ustawy, o której mowa w art. 13, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 38 ust. 7 ustawy, o której mowa w art. 13, w brzmieniu nadanym niniejszą ustawą,
9) art. 30c ustawy, o której mowa w art. 5, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 30zb ustawy, o której mowa w art. 5, w brzmieniu nadanym niniejszą ustawą,
10) art. 89 ust. 7, art. 89a ust. 8 i art. 89b ust. 2 ustawy, o której mowa w art. 6, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 107zc ustawy, o której mowa w art. 6, w brzmieniu nadanym niniejszą ustawą,
11) art. 89e ust. 3 ustawy, o której mowa w art. 6, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 107zf ust. 4 ustawy, o której mowa w art. 6, w brzmieniu nadanym niniejszą ustawą,
11a) art. 56 ust. 3 ustawy, o której mowa w art. 7, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 56 ust. 3 ustawy, o której mowa w art. 7, w brzmieniu nadanym niniejszą ustawą,
12) art. 192 ust. 2 ustawy, o której mowa w art. 7, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 192 ust. 2 ustawy, o której mowa w art. 7, w brzmieniu nadanym niniejszą ustawą,
13) art. 12 ust. 2 i art. 26 ust. 4 ustawy, o której mowa w art. 9, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 12e i art. 26 ust. 5 ustawy, o której mowa w art. 9, w brzmieniu nadanym niniejszą ustawą,
14) art. 27 ust. 9 i art. 29 ust. 7 ustawy, o której mowa w art. 11, zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 27 ust. 9 i art. 29 ust. 7 ustawy, o której mowa w art. 11, w brzmieniu nadanym niniejszą ustawą
– nie dłużej jednak niż do dnia 1 stycznia 2018 r.”
„Art. 50. 1. System monitorowania programów lekowych, o którym mowa w ustawie, o której mowa w art. 13, funkcjonujący w dniu wejścia w życie niniejszej ustawy staje się, z tym dniem, systemem monitorowania programów lekowych, o którym mowa w art. 188c ustawy, o której mowa w art. 7, w brzmieniu nadanym niniejszą ustawą.
2. Prezes Narodowego Funduszu Zdrowia w terminie 18 miesięcy od dnia wejścia w życie niniejszej ustawy dostosuje system monitorowania programów lekowych, o których mowa w art. 188c ustawy, o której mowa w art. 7, w brzmieniu nadanym niniejszą ustawą, do programów lekowych, o których mowa w ustawie, o której mowa w art. 13, nieobjętych tym systemem w dniu wejścia w życie tej ustawy.”
„Art. 52. Ustawa wchodzi w życie po upływie 14 dni od dnia ogłoszenia, z wyjątkiem:
1) art. 6 pkt 8, który wchodzi w życie z dniem 1 stycznia 2016 r.;
2) art. 6 pkt 7 lit. b i c oraz pkt 13 lit. c i d, które wchodzą w życie z dniem 1 kwietnia 2016 r.;
3) art. 3 pkt 2, art. 5, art. 6 pkt 16 i art. 29–44, które wchodzą w życie z dniem 1 maja 2016 r.;
4) art. 1 pkt 15 w zakresie art. 17a ust. 2 pkt 4 i art. 6 pkt 1, które wchodzą w życie z dniem 1 lipca 2016 r.;
5) art. 1 pkt 28, art. 6 pkt 3 i art. 13 pkt 2–10, które wchodzą w życie z dniem 1 stycznia 2017 r.;
5a) art. 4 pkt 2 lit. b i pkt 3–32, art. 9 pkt 1–4, 10 i 11, art. 15 pkt 8–17 i art. 19–28, które wchodzą w życie z dniem 1 maja 2017 r.;
6) art. 7 pkt 23 w zakresie art. 188c ust. 3, ust. 4 pkt 2 i ust. 6, które wchodzą w życie z dniem 1 lipca 2017 r.;
7) art. 9 pkt 7 i 8, które wchodzą w życie z dniem 1 sierpnia 2017 r.”;
3) art. 5–8 ustawy z dnia 18 marca 2016 r. o zmianie ustawy o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych oraz niektórych innych ustaw (Dz. U. poz. 652), które stanowią:
„Art. 5. Pierwszy wykaz, o którym mowa w art. 37 ust. 1 ustawy zmienianej w art. 2, w części dotyczącej bezpłatnego zaopatrzenia świadczeniobiorców po ukończeniu 75. roku życia w leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne, minister właściwy do spraw zdrowia ogłosi nie później niż do dnia 1 września 2016 r., biorąc pod uwagę wytyczne określone w art. 43a ust. 2 pkt 1–3 ustawy, o której mowa w art. 1, w brzmieniu nadanym niniejszą ustawą.
Art. 6. Do postępowań w sprawie wniosków, o których mowa w art. 24 ust. 1 pkt 1 ustawy zmienianej w art. 2, wszczętych i niezakończonych przed dniem wejścia w życie niniejszej ustawy stosuje się przepisy dotychczasowe.
Art. 7. 1. W latach 2016–2025 maksymalny limit wydatków z budżetu państwa, będących konsekwencją wejścia ustawy w życie, wynosi 8 274 600 tys. zł, przy czym w kolejnych latach wyniesie:
1) 2016 r. – 125 000 tys. zł;
2) 2017 r. – 564 300 tys. zł;
3) 2018 r. – 643 300 tys. zł;
4) 2019 r. – 733 400 tys. zł;
5) 2020 r. – 836 000 tys. zł;
6) 2021 r. – 953 100 tys. zł;
7) 2022 r. – 1 010 300 tys. zł;
8) 2023 r. – 1 070 900 tys. zł;
9) 2024 r. – 1 135 100 tys. zł;
10) 2025 r. – 1 203 200 tys. zł.
2. Minister właściwy do spraw zdrowia monitoruje co najmniej w okresach miesięcznych wykorzystanie rocznych limitów wydatków, o których mowa w ust. 1.
3. W przypadku gdy:
1) może nastąpić przekroczenie limitu wydatków, o których mowa w ust. 1, przewidzianych w 2016 r. lub
2) wydatki, o których mowa w ust. 1 pkt 1, przekroczą po pierwszych dwóch miesiącach od dnia obowiązywania wykazu, o którym mowa w art. 37 ust. 1 ustawy zmienianej w art. 2, w części dotyczącej bezpłatnego zaopatrzenia świadczeniobiorców po ukończeniu 75. roku życia w leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne, 50% limitu przewidzianego na ten rok
– minister właściwy do spraw zdrowia wdraża mechanizm korygujący, polegający na zmianie dotyczącej wykazu leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych, o którym mowa w art. 37 ust. 1 ustawy zmienianej w art. 2, w zakresie leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych, wydawanych bezpłatnie świadczeniobiorcom po ukończeniu 75. roku życia.
4. W przypadku gdy:
1) może nastąpić przekroczenie limitu wydatków, o których mowa w ust. 1, przewidzianych na okres jednego roku lub
2) wydatki, o których mowa w ust. 1 pkt 2–10, przekroczą w kwartale danego roku 25% limitu przewidzianego na ten rok – minister właściwy do spraw zdrowia wdraża mechanizm korygujący, polegający na zmianie dotyczącej wykazu leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych, o którym mowa w art. 37 ust. 1 ustawy zmienianej w art. 2, w zakresie leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych, wydawanych bezpłatnie świadczeniobiorcom po ukończeniu 75. roku życia.
Art. 8. Ustawa wchodzi w życie po upływie 30 dni od dnia ogłoszenia, z wyjątkiem art. 4 pkt 1, który wchodzi w życie z dniem 1 stycznia 2017 r.”.
Marszałek Sejmu: M. Kuchciński
Załącznik do obwieszczenia Marszałka Sejmu Rzeczypospolitej Polskiej
z dnia 23 sierpnia 2016 r. (poz. 1536)
USTAWA
z dnia 12 maja 2011 r.
o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych1)
Rozdział 1
Przepisy ogólne
Art. 1. Ustawa określa:
1) zasady, warunki i tryb podejmowania decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego i uchylenia tej decyzji;
2) zasady finansowania leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego objętych decyzją, o której mowa w pkt 1;
3) kryteria tworzenia poziomów odpłatności i grup limitowych leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych objętych decyzją, o której mowa w pkt 1;
4) zasady i tryb oraz kryteria ustalania urzędowych cen zbytu na leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne, a także wysokość urzędowych marż hurtowych i urzędowych marż detalicznych;
5) zasady ustalania cen leków oraz środków spożywczych specjalnego przeznaczenia żywieniowego stosowanych w ramach udzielania świadczeń gwarantowanych;
6) zasady finansowania ze środków publicznych wyrobów medycznych przysługujących świadczeniobiorcom na zlecenie osoby uprawnionej;
7) obowiązki aptek wynikające z obrotu lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi, objętymi decyzją, o której mowa w pkt 1, a także zasady kontroli aptek;
8) obowiązki osób uprawnionych do wystawiania recept na leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne, objęte decyzją, o której mowa w pkt 1.
Art. 2. Użyte w ustawie określenia oznaczają:
1) Agencja – Agencję Oceny Technologii Medycznych i Taryfikacji działającą na podstawie ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych (Dz. U. z 2015 r. poz. 581, z późn. zm.2));
2) apteka – aptekę ogólnodostępną lub punkt apteczny w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne (Dz. U. z 2008 r. poz. 271, z późn. zm.3));
3) całkowity budżet na refundację – wysokość środków publicznych przeznaczonych w planie finansowym Narodowego Funduszu Zdrowia, o którym mowa w art. 118 ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych, na refundowane leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne, o których mowa w art. 15 ust. 2 pkt 14, 16–18 oraz objęte programami lekowymi, o których mowa w art. 15 ust. 2 pkt 15 tej ustawy;
4) cena detaliczna – urzędową cenę zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego powiększoną o urzędową marżę hurtową i urzędową marżę detaliczną oraz należny podatek od towarów i usług;
5) cena hurtowa – urzędową cenę zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego powiększoną o urzędową marżę hurtową oraz należny podatek od towarów i usług;
6) cena zbytu netto – cenę sprzedaży leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego do podmiotów uprawnionych do obrotu, nieuwzględniającą należnego podatku od towarów i usług;
7) DDD – dobową dawkę leku ustaloną przez Światową Organizację Zdrowia;
8) Fundusz – Narodowy Fundusz Zdrowia w rozumieniu ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych;
9) grupa limitowa – grupę leków albo środków spożywczych specjalnego przeznaczenia żywieniowego albo wyrobów medycznych objętych wspólnym limitem finansowania;
10) lek – produkt leczniczy w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne;
11) lek recepturowy – lek sporządzony w aptece na podstawie recepty lekarskiej;
12) nazwa międzynarodowa leku – nazwę leku zalecaną przez Światową Organizację Zdrowia;
13) odpowiednik – w przypadku:
a) leku – lek zawierający tę samą substancję czynną oraz mający te same wskazania i tę samą drogę podania przy braku różnic postaci farmaceutycznej,
b) środka spożywczego specjalnego przeznaczenia żywieniowego – środek spożywczy specjalnego przeznaczenia żywieniowego mający ten sam albo zbliżony skład, zastosowanie lub sposób przygotowania,
c) wyrobu medycznego – wyrób medyczny mający takie samo przewidziane zastosowanie oraz właściwości;
14)4) osoba uprawniona – osobę posiadającą prawo wykonywania zawodu medycznego, która na podstawie przepisów dotyczących wykonywania danego zawodu medycznego, jest uprawniona do wystawiania recept zgodnie z ustawą oraz ustawą z dnia 6 września 2001 r. – Prawo farmaceutyczne oraz zleceń na zaopatrzenie w wyroby medyczne, o których mowa w art. 38;
15) podmiot działający na rynku spożywczym – podmiot działający na rynku spożywczym w rozumieniu art. 3 pkt 3 rozporządzenia (WE) nr 178/2002 Parlamentu Europejskiego i Rady z dnia 28 stycznia 2002 r. ustanawiającego ogólne zasady i wymagania prawa żywnościowego, powołującego Europejski Urząd do Spraw Bezpieczeństwa Żywności oraz ustanawiającego procedury w sprawie bezpieczeństwa żywności (Dz. Urz. UE L 179 z 07.07.2007, str. 59);
16) podmiot odpowiedzialny – podmiot odpowiedzialny w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne;
17) podmiot zobowiązany do finansowania świadczeń ze środków publicznych – podmiot zobowiązany do finansowania świadczeń opieki zdrowotnej ze środków publicznych w rozumieniu przepisów ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych;
18) program lekowy – program zdrowotny w rozumieniu przepisów ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych obejmujący technologię lekową, w której substancja czynna nie jest składową kosztową innych świadczeń gwarantowanych lub środek spożywczy specjalnego przeznaczenia żywieniowego, który nie jest składową kosztową innych świadczeń gwarantowanych w rozumieniu tej ustawy;
19) przedsiębiorca – przedsiębiorcę w rozumieniu ustawy z dnia 2 lipca 2004 r. o swobodzie działalności gospodarczej (Dz. U. z 2015 r. poz. 584, z późn. zm.5));
20) Rada Przejrzystości – Radę Przejrzystości działającą na podstawie ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych;
21) środek spożywczy specjalnego przeznaczenia żywieniowego – środek spożywczy, o którym mowa w art. 24 ust. 2 pkt 1 i 4 ustawy z dnia 25 sierpnia 2006 r. o bezpieczeństwie żywności i żywienia (Dz. U. z 2015 r. poz. 594 i 1893 oraz z 2016 r. poz. 65) przeznaczony do dietetycznego odżywiania pacjentów pod nadzorem lekarza, którego stosowania nie można uniknąć przez modyfikację normalnej diety lub podawanie innych środków spożywczych specjalnego przeznaczenia żywieniowego;
22) świadczenie gwarantowane – świadczenie gwarantowane w rozumieniu ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych;
23) świadczeniobiorca – świadczeniobiorcę w rozumieniu ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych;
24) świadczeniodawca – świadczeniodawcę w rozumieniu ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych;
25) technologia lekowa – technologię medyczną w rozumieniu ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych, której główną składową kosztową jest lek;
26) urzędowa cena zbytu – cenę zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego ustaloną w decyzji administracyjnej o objęciu refundacją, uwzględniającą należny podatek od towarów i usług;
27) wnioskodawca – podmiot odpowiedzialny, przedstawiciela podmiotu odpowiedzialnego, podmiot uprawniony do importu równoległego w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, wytwórcę wyrobów medycznych, jego autoryzowanego przedstawiciela, dystrybutora albo importera, w rozumieniu ustawy z dnia 20 maja 2010 r. o wyrobach medycznych (Dz. U. z 2015 r. poz. 876 i 1918 oraz z 2016 r. poz. 542), a także podmiot działający na rynku spożywczym;
28) wyrób medyczny – wyrób medyczny, wyposażenie wyrobu medycznego, wyrób medyczny do diagnostyki in vitro, wyposażenie wyrobu medycznego do diagnostyki in vitro, o których mowa w art. 2 ust. 1 pkt 33, 34, 38 i 39 ustawy z dnia 20 maja 2010 r. o wyrobach medycznych;
29) wytwórca wyrobu medycznego – wytwórcę w rozumieniu ustawy z dnia 20 maja 2010 r. o wyrobach medycznych.
Art. 3. 1. Całkowity budżet na refundację wynosi nie więcej niż 17% sumy środków publicznych przeznaczonych na finansowanie świadczeń gwarantowanych w planie finansowym Funduszu.
2. Kwotę środków finansowych stanowiącą wzrost całkowitego budżetu na refundację w roku rozliczeniowym w stosunku do całkowitego budżetu na refundację w roku poprzedzającym przeznacza się na:
1) finansowanie:
a) dotychczas nieobjętych refundacją leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, które nie mają swojego odpowiednika refundowanego w danym wskazaniu, z zakresu, o którym mowa w art. 15 ust. 2 pkt 14–16 ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych, zwanej dalej „ustawą o świadczeniach”, wobec których została wydana decyzja administracyjna o objęciu refundacją,
b) przewidywanego wzrostu refundacji w wybranych grupach limitowych wynikającego ze zmian w Charakterystyce Produktu Leczniczego lub ze zmian praktyki klinicznej;
2) refundację, w części dotyczącej finansowania świadczeń, o których mowa w art. 15 ust. 2 pkt 14 ustawy o świadczeniach.
3. Kwota środków finansowych, o której mowa w ust. 2 pkt 1, stanowi rezerwę.
4. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, podział kwoty środków finansowych, o której mowa w ust. 2, uwzględniając realizację refundacji, a także liczbę i rodzaj złożonych wniosków, o których mowa w art. 24 ust. 1.
Art. 4. 1. W przypadku gdy w trakcie realizacji planu finansowego Funduszu dojdzie do przekroczenia całkowitego budżetu na refundację, w części dotyczącej finansowania świadczeń, o których mowa w art. 15 ust. 2 pkt 14 ustawy o świadczeniach, wyznacza się kwotę przekroczenia dla danej grupy limitowej. Wnioskodawca, który uzyskał decyzję administracyjną o objęciu refundacją, zwraca do Funduszu kwotę proporcjonalną do udziału kosztów refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego objętego tą decyzją, w tym przekroczeniu, w danej grupie limitowej.
2. Kwotę przekroczenia w danej grupie limitowej oblicza się według wzoru:
KP = Wr' – Wr
gdzie poszczególne symbole oznaczają:
KP – kwotę przekroczenia,
Wr' – kwotę refundacji w roku rozliczeniowym w danej grupie limitowej,
Wr – planowaną kwotę refundacji w danej grupie limitowej, wyliczoną jako iloczyn planowanej kwoty refundacji w tej grupie w roku poprzedzającym i współczynnika wzrostu stanowiącego iloraz całkowitego budżetu na refundację w roku rozliczeniowym pomniejszonego o rezerwę, o której mowa w art. 3 ust. 3, i całkowitego budżetu na refundację w roku poprzedzającym.
3. Udział w zwrocie kwoty przekroczenia w danej grupie limitowej biorą jedynie ci wnioskodawcy, którzy otrzymali decyzję administracyjną o objęciu refundacją, dla których dynamika poziomu refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego w danej grupie limitowej, w roku rozliczeniowym względem roku poprzedzającego, jest równa albo większa od 1, zgodnie z poniższym wzorem:
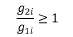
gdzie poszczególne symbole oznaczają:
g1i – kwotę refundacji na koniec roku poprzedzającego dla produktu i-tego wnioskodawcy, który otrzymał decyzję administracyjną o objęciu refundacją w danej grupie limitowej,
g2i – kwotę refundacji na koniec roku rozliczeniowego dla produktu i-tego wnioskodawcy, który otrzymał decyzję administracyjną o objęciu refundacją w danej grupie limitowej.
4. W przypadku wnioskodawcy, który otrzymał decyzję administracyjną o objęciu refundacją, a który nie uzyskiwał przychodów z tytułu refundacji w danej grupie limitowej w roku poprzedzającym, współczynnik dynamiki poziomu refundacji w danej grupie limitowej przyjmuje wartość 1.
5. Udział w kwocie przekroczenia jest proporcjonalny do udziału kwoty refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, w sumarycznej kwocie refundacji produktów w danej grupie limitowej, o których mowa w ust. 3. Udział w kwocie przekroczenia jest korygowany o iloraz urzędowej ceny zbytu za DDD leku wnioskodawcy, który otrzymał decyzję administracyjną o objęciu refundacją, na koniec roku rozliczeniowego i najniższej urzędowej ceny zbytu za DDD leku stanowiącego podstawę limitu w danej grupie limitowej w roku rozliczeniowym. Unormowany współczynnik udziału w kwocie przekroczenia w danej grupie limitowej dla produktu refundowanego danego wnioskodawcy, który otrzymał decyzję administracyjną o objęciu refundacją, wyliczany jest według wzoru:
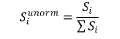
gdzie:
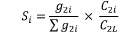
gdzie poszczególne symbole oznaczają:
Siunorm – unormowany współczynnik udziału w kwocie przekroczenia dla produktu i-tego wnioskodawcy, który otrzymał decyzję o objęciu refundacją w danej grupie limitowej,
Si – współczynnik udziału w kwocie przekroczenia dla produktu i-tego wnioskodawcy, który otrzymał decyzję o objęciu refundacją w danej grupie limitowej,
g2i – kwotę refundacji na koniec roku rozliczeniowego dla produktu i-tego wnioskodawcy, który otrzymał decyzję o objęciu refundacją w danej grupie limitowej,
∑g2i – sumaryczną kwotę refundacji na koniec roku rozliczeniowego produktów wszystkich wnioskodawców, którzy otrzymali decyzję administracyjną o objęciu refundacją w danej grupie limitowej,
C2i – urzędową cenę zbytu za DDD leku i-tego wnioskodawcy, który otrzymał decyzję administracyjną o objęciu refundacją w danej grupie limitowej na koniec roku rozliczeniowego,
C2L – najniższą urzędową cenę zbytu za DDD leku stanowiącego podstawę limitu w danej grupie limitowej w roku rozliczeniowym.
6. Przepis ust. 5 stosuje się odpowiednio do środka spożywczego specjalnego przeznaczenia żywieniowego i wyrobu medycznego oraz leku, w odniesieniu do którego nie określono DDD.
7. Wnioskodawca, który otrzymał decyzję administracyjną o objęciu refundacją, biorący udział w zwrocie kwoty przekroczenia, zwraca Funduszowi kwotę odpowiednio dla danej grupy limitowej, w wysokości wyliczanej według wzoru:
KZi = Siunorm × KP × G × 0,5
gdzie poszczególne symbole oznaczają:
KZi – kwotę zwracaną przez i-tego wnioskodawcę, który otrzymał decyzję administracyjną o objęciu refundacją w danej grupie limitowej,
Siunorm – unormowany współczynnik udziału w kwocie przekroczenia dla i-tego wnioskodawcy, który otrzymał decyzję administracyjną o objęciu refundacją,
KP – kwotę przekroczenia w danej grupie limitowej,
G – współczynnik korygujący stanowiący iloraz różnicy poniesionych wydatków na refundację leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych w części dotyczącej finansowania świadczeń, o których mowa w art. 15 ust. 2 pkt 14 ustawy o świadczeniach, w danym roku rozliczeniowym i całkowitego budżetu na refundację w tej części w tym roku oraz sumy kwot przekroczenia w grupach limitowych.
8. Kwotę przekroczenia oraz kwotę zwrotu oblicza Fundusz w terminie 30 dni od zatwierdzenia sprawozdania finansowego Funduszu za rok poprzedni na podstawie danych, o których mowa w art. 45 ust. 1.
9. Zestawienie kwot zwrotu w odniesieniu do poszczególnych grup limitowych dla każdego produktu objętego refundacją, obliczonych w sposób określony w ust. 7, Prezes Funduszu niezwłocznie przekazuje ministrowi właściwemu do spraw zdrowia.
10. Kwotę zwrotu ustala w drodze decyzji administracyjnej minister właściwy do spraw zdrowia i podlega ona uiszczeniu w terminie 30 dni od dnia, w którym decyzja stała się ostateczna.
11. Przepisów ust. 1–10 nie stosuje się w przypadku ustalenia w decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, instrumentów dzielenia ryzyka, o których mowa w art. 11 ust. 5.
Art. 5. W przypadku, gdy lek zawiera więcej niż jedną substancję czynną za podstawę obliczeń, o których mowa w art. 4, 6, 7, 9 i art. 13–15, przyjmuje się cenę DDD lub liczbę DDD substancji czynnej zawartej w tym leku o najwyższym koszcie DDD.
Rozdział 2
Poziomy odpłatności i marże refundowanych leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych
Art. 6. 1. Ustala się kategorię dostępności refundacyjnej:
1) lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny dostępny w aptece na receptę:
a) w całym zakresie zarejestrowanych wskazań i przeznaczeń,
b) we wskazaniu określonym stanem klinicznym;
2) lek, środek spożywczy specjalnego przeznaczenia żywieniowego stosowany w ramach programu lekowego;
3) lek stosowany w ramach chemioterapii:
a) w całym zakresie zarejestrowanych wskazań i przeznaczeń,
b) we wskazaniu określonym stanem klinicznym;
4) lek, środek spożywczy specjalnego przeznaczenia żywieniowego stosowany w ramach udzielania świadczeń gwarantowanych, innych niż wymienione w pkt 1–3.
2. Lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny, dla którego wydana została decyzja administracyjna o objęciu refundacją w zakresie nadanej kategorii dostępności refundacyjnej, o której mowa w ust. 1 pkt 1, jest wydawany świadczeniobiorcy:
1) bezpłatnie,
2) za odpłatnością ryczałtową,
3) za odpłatnością w wysokości 30% albo 50% ich limitu finansowania
– do wysokości limitu finansowania i za dopłatą w wysokości różnicy między ceną detaliczną a wysokością limitu finansowania.
3. Odpłatności, o których mowa w ust. 2, dotyczą jednostkowego opakowania leku, środka spożywczego specjalnego przeznaczenia żywieniowego oraz jednostkowego wyrobu medycznego, z tym że odpłatność, o której mowa w ust. 2 pkt 2, dotyczy jednostkowego opakowania leku zawierającego nie więcej niż 30 DDD, a w przypadku większej liczby DDD w opakowaniu odpłatność ta zwiększana jest proporcjonalnie do ilorazu liczby DDD w opakowaniu i 30 DDD.
4. W przypadku leku, dla którego DDD nie zostało określone, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego odpłatność, o której mowa w ust. 2 pkt 2, dotyczy jednostkowego opakowania leku albo środka spożywczego specjalnego przeznaczenia żywieniowego, albo liczby jednostkowych wyrobów medycznych, albo liczby jednostek wyrobu medycznego wystarczających na miesięczną terapię lub stosowanie. Zasady zwiększania odpłatności, o której mowa w ust. 2 pkt 2, określone w ust. 3 stosuje się odpowiednio.
5. Leki recepturowe przygotowane z surowców farmaceutycznych lub z leków gotowych, dla których została wydana decyzja administracyjna o objęciu refundacją, są wydawane świadczeniobiorcy za odpłatnością ryczałtową, pod warunkiem, że przepisana dawka leku recepturowego jest mniejsza od najmniejszej dawki leku gotowego w formie stałej stosowanej doustnie.
6. Odpłatność ryczałtowa, o której mowa w ust. 2 pkt 2, wynosi 3,20 zł.
7. Odpłatność ryczałtowa, o której mowa w ust. 5, wynosi 0,50% wysokości minimalnego wynagrodzenia za pracę ogłaszanego w obwieszczeniu Prezesa Rady Ministrów wydanym na podstawie art. 2 ust. 4 ustawy z dnia 10 października 2002 r. o minimalnym wynagrodzeniu za pracę (Dz. U. z 2015 r. poz. 2008), z zaokrągleniem do pierwszego miejsca po przecinku.
8. Lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny, dla którego wydana została decyzja administracyjna o objęciu refundacją w zakresie nadanej kategorii dostępności refundacyjnej, o której mowa w ust. 1 pkt 2 i 3, jest wydawany świadczeniobiorcy bezpłatnie.
9. Apteka zobowiązana jest stosować odpłatność wynikającą z ustawy.
10. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia:
1) wykaz leków, które mogą być traktowane jako surowce farmaceutyczne przy sporządzaniu leków recepturowych,
2) ilość leku recepturowego, którego dotyczy odpłatność ryczałtowa, oraz sposób obliczania kosztu sporządzania leku recepturowego
– biorąc pod uwagę dostępność do leków, bezpieczeństwo ich stosowania oraz postać farmaceutyczną.
Art. 7. 1. Ustala się urzędową marżę hurtową w wysokości 5% urzędowej ceny zbytu.
2. Przedsiębiorcy prowadzący obrót hurtowy, w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, są obowiązani stosować marżę, o której mowa w ust. 1.
3. Marża, o której mowa w ust. 1, może być dzielona pomiędzy przedsiębiorców prowadzących obrót hurtowy.
4. Ustala się urzędową marżę detaliczną naliczaną od ceny hurtowej leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo wyrobu medycznego stanowiącego podstawę limitu w danej grupie limitowej, w wysokości:
| od | do | zasada marży |
| – | 5,00 zł | 40% |
| 5,01 zł | 10,00 zł | 2 zł + 30% × (x – 5,00 zł) |
| 10,01 zł | 20,00 zł | 3,50 zł + 20% × (x – 10,00 zł) |
| 20,01 zł | 40,00 zł | 5,50 zł + 15% × (x – 20,00 zł) |
| 40,01 zł | 80,00 zł | 8,50 zł + 10% × (x – 40,00 zł) |
| 80,01 zł | 160,00 zł | 12,50 zł + 5% × (x – 80,00 zł) |
| 160,01 zł | 320,00 zł | 16,50 zł + 2,5% × (x – 160,00 zł) |
| 320,01 zł | 640,00 zł | 20,50 zł + 2,5% × (x – 320,00 zł) |
| 640,01 zł | 1280,00 zł | 28,50 zł + 2,5% × (x – 640,00 zł) |
| 1280,01 zł |
| 44,50 zł + 1,25% × (x – 1280,00 zł) |
– gdzie x oznacza cenę hurtową leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo wyrobu medycznego stanowiącego podstawę limitu, uwzględniającą liczbę DDD leku, jednostek środka spożywczego specjalnego przeznaczenia żywieniowego w opakowaniu albo liczbę jednostkowych wyrobów medycznych, albo liczbę jednostek wyrobu medycznego.
5. Podmioty uprawnione do obrotu detalicznego, w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, są obowiązane stosować marżę, o której mowa w ust. 4.
6. Dla leków wydawanych w trybie, o którym mowa w art. 46 ustawy o świadczeniach, dla których nie ustalono limitu finansowania, stosuje się urzędową marżę detaliczną, w wysokości określonej w ust. 4, liczoną od ceny hurtowej, i nie może być wyższa niż 20 zł.
7. Dla leków oraz środków spożywczych specjalnego przeznaczenia żywieniowego, wydawanych w trybie, o którym mowa w art. 39 ust. 1, ustala się marżę hurtową w wysokości 10%. Marża detaliczna wynosi 100% wartości urzędowej marży detalicznej ustalonej w ust. 4 dla danego przedziału ceny hurtowej.
8. Dla leków, o których mowa w art. 6 ust. 5, ustala się marżę detaliczną w wysokości 25% liczoną od kosztu jego sporządzenia.
Art. 8. Urzędowe ceny zbytu, a także urzędowe marże hurtowe i detaliczne, mają charakter cen i marż sztywnych.
Art. 9. 1. Świadczeniodawca w celu realizacji świadczeń gwarantowanych jest obowiązany nabywać leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne po cenie nie wyższej niż urzędowa cena zbytu powiększona o marżę nie wyższą niż urzędowa marża hurtowa, a w przypadku nabywania od podmiotu innego niż przedsiębiorca prowadzący obrót hurtowy w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne – po cenie nie wyższej niż urzędowa cena zbytu.
2. Lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny, o którym mowa w art. 6 ust. 1 pkt 1–3, świadczeniodawca jest obowiązany nabywać po cenie nie wyższej niż urzędowa cena zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, stanowiącego podstawę limitu, uwzględniając liczbę DDD leku, jednostek środka spożywczego specjalnego przeznaczenia żywieniowego w opakowaniu albo liczbę jednostkowych wyrobów medycznych, albo liczbę jednostek wyrobu medycznego, powiększoną o marżę nie wyższą niż urzędowa marża hurtowa, a w przypadku nabywania od podmiotu innego niż przedsiębiorca prowadzący obrót hurtowy w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne – po cenie nie wyższej niż urzędowa cena zbytu.
3. Dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, o którym mowa w art. 6 ust. 1 pkt 4, urzędową cenę zbytu ustala się:
1) na wniosek;
2) z urzędu, w przypadku gdy stanowią one istotną składową kosztową świadczeń gwarantowanych.
4. Minister właściwy do spraw zdrowia, wszczynając postępowanie z urzędu, wzywa podmiot odpowiedzialny lub podmiot działający na rynku spożywczym do przedstawienia informacji określonych w art. 28 pkt 3–7.
5. Podmiot, o którym mowa w ust. 4, jest zobowiązany do przedstawienia informacji, o których mowa w art. 28 pkt 1–7, w terminie 60 dni od dnia doręczenia wezwania.
6. W przypadku nieprzedstawienia informacji, o których mowa w art. 28 pkt 3–7, minister właściwy do spraw zdrowia ustala urzędową cenę zbytu na podstawie kryteriów, o których mowa w art. 13 ust. 8.
7. W przypadku, o którym mowa w ust. 3 pkt 2, opłat, o których mowa w art. 32 ust. 1, nie pobiera się.
Rozdział 3
Kryteria tworzenia poziomów odpłatności i grup limitowych oraz kryteria podejmowania decyzji o objęciu refundacją i zasady ustalania urzędowej ceny zbytu
Art. 10. 1. Refundowany może być lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny, który spełnia następujące wymagania:
1) jest dopuszczony do obrotu lub pozostaje w obrocie w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, albo jest wprowadzony do obrotu i do używania w rozumieniu ustawy z dnia 20 maja 2010 r. o wyrobach medycznych, albo jest wprowadzony do obrotu w rozumieniu ustawy z dnia 25 sierpnia 2006 r. o bezpieczeństwie żywności i żywienia;
2) jest dostępny na rynku;
3) posiada kod identyfikacyjny EAN lub inny kod odpowiadający kodowi EAN.
2. Refundowany może być również:
1) lek nieposiadający pozwolenia na dopuszczenie do obrotu na terytorium Rzeczypospolitej Polskiej i sprowadzany z zagranicy na warunkach i w trybie art. 4 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne;
2) lek, o którym mowa w art. 40;
3) środek spożywczy specjalnego przeznaczenia żywieniowego, o którym mowa w art. 29a ustawy z dnia 25 sierpnia 2006 r. o bezpieczeństwie żywności i żywienia.
3. Refundowany nie może być:
1) lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny w stanach klinicznych, w których możliwe jest skuteczne zastąpienie tego leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego poprzez zmianę stylu życia pacjenta;
2) lek o kategorii dostępności Rp, który posiada swój odpowiednik o kategorii dostępności OTC, chyba że wymaga stosowania dłużej niż 30 dni w określonym stanie klinicznym;
3) lek, środek spożywczy specjalnego przeznaczenia żywieniowego, ujęty w wykazie określonym w przepisach wydanych na podstawie art. 39 ust. 5.
Art. 11. 1. Objęcie refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego następuje w drodze decyzji administracyjnej ministra właściwego do spraw zdrowia.
2. Decyzja, o której mowa w ust. 1, zawiera:
1) oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej;
2) nazwę leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo nazwę i zastosowanie wyrobu medycznego, oraz jego dane identyfikujące;
3) kategorię dostępności refundacyjnej, a w przypadku kategorii, o której mowa w art. 6 ust. 1 pkt 2 – opis programu lekowego stanowiący załącznik do decyzji;
4) poziom odpłatności;
5) urzędową cenę zbytu;
6) termin wejścia w życie decyzji oraz okres jej obowiązywania;
7) instrumenty dzielenia ryzyka, jeżeli zostały ustalone;
8) określenie grupy limitowej.
3. Decyzję, o której mowa w ust. 1, wydaje się na okres:
1) 5 lat – dla leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, dla których nieprzerwanie obowiązywała decyzja administracyjna o objęciu refundacją lub w stosunku do których decyzja dla odpowiednika refundowanego w ramach tej samej kategorii dostępności refundacyjnej i w tym samym wskazaniu obowiązywała nieprzerwanie, przez okres dłuższy niż 5 lat,
2) 3 lat – dla leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, dla których nieprzerwanie obowiązywała decyzja administracyjna o objęciu refundacją lub w stosunku do których decyzja dla odpowiednika refundowanego w ramach tej samej kategorii dostępności refundacyjnej i w tym samym wskazaniu obowiązywała nieprzerwanie, przez okres od 3 do 5 lat,
3) 2 lat – dla leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, dla których nieprzerwanie obowiązywała decyzja administracyjna o objęciu refundacją lub w stosunku do których decyzja dla odpowiednika refundowanego w ramach tej samej kategorii dostępności refundacyjnej i w tym samym wskazaniu obowiązywała nieprzerwanie, przez okres krótszy niż 3 lata, albo dla których wydawana jest pierwsza decyzja administracyjna o objęciu refundacją
– przy czym okres obowiązywania decyzji nie może przekraczać terminu wygaśnięcia okresu wyłączności rynkowej.
4. Podwyższenie albo obniżenie urzędowej ceny zbytu następuje w drodze zmiany decyzji, o której mowa w ust. 1.
5. Instrumenty dzielenia ryzyka, o których mowa w ust. 2 pkt 7, mogą dotyczyć:
1) uzależnienia wielkości przychodu wnioskodawcy od uzyskiwanych efektów zdrowotnych;
2) uzależnienia wysokości urzędowej ceny zbytu od zapewnienia przez wnioskodawcę dostaw po obniżonej ustalonej w negocjacjach cenie leku, środka spożywczego specjalnego przeznaczenia żywieniowego oraz wyrobu medycznego;
3) uzależnienia wysokości urzędowej ceny zbytu od wielkości obrotu lekiem, środkiem spożywczym specjalnego przeznaczenia żywieniowego oraz wyrobem medycznym;
4) uzależnienia wysokości urzędowej ceny zbytu od zwrotu części uzyskanej refundacji podmiotowi zobowiązanemu do finansowania świadczeń ze środków publicznych;
5) ustalenia innych warunków refundacji mających wpływ na zwiększenie dostępności do świadczeń gwarantowanych lub obniżenie kosztów tych świadczeń.
6. Minister właściwy do spraw zdrowia, w drodze decyzji administracyjnej, ustala urzędową cenę zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, o którym mowa w art. 6 ust. 1 pkt 4.
7. Decyzja, o której mowa w ust. 6, zawiera:
1) oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej;
2) nazwę leku, środka spożywczego specjalnego przeznaczenia żywieniowego oraz jego dane identyfikujące;
3) urzędową cenę zbytu.
8. Okres obowiązywania decyzji, o której mowa w ust. 6, wynosi 5 lat.
9. Decyzja, o której mowa w ust. 6, wygasa w dniu umieszczenia leku, środka specjalnego przeznaczenia żywieniowego w obwieszczeniu, o którym mowa w art. 37. Wygaśnięcie stwierdza minister do spraw zdrowia w drodze decyzji.
10. Skrócenie okresu obowiązywania decyzji, o którym mowa w ust. 3 albo ust. 8, następuje w drodze decyzji ministra właściwego do spraw zdrowia.
11. Minister właściwy do spraw zdrowia odmawia wydania decyzji, o której mowa w ust. 10, jeżeli:
1) jej wydanie spowodowałoby:
a) istotne ograniczenie dostępności świadczeniobiorców do leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych,
b) znaczny wzrost odpłatności lub dopłat świadczeniobiorców;
2) urzędowa cena zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, o którym mowa w art. 6 ust. 1 pkt 4, została ustalona z urzędu.
Art. 12. Minister właściwy do spraw zdrowia, mając na uwadze uzyskanie jak największych efektów zdrowotnych w ramach dostępnych środków publicznych, wydaje decyzję administracyjną o objęciu refundacją i ustaleniu urzędowej ceny zbytu, przy uwzględnieniu następujących kryteriów:
1) stanowiska Komisji Ekonomicznej, o której mowa w art. 17,
2) rekomendacji Prezesa Agencji, o której mowa w art. 35 ust. 6,
3) istotności stanu klinicznego, którego dotyczy wniosek o objęcie refundacją,
4) skuteczności klinicznej i praktycznej,
5) bezpieczeństwa stosowania,
6) relacji korzyści zdrowotnych do ryzyka stosowania,
7) stosunku kosztów do uzyskiwanych efektów zdrowotnych dotychczas refundowanych leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, w porównaniu z wnioskowanym,
8) konkurencyjności cenowej,
9) wpływu na wydatki podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych i świadczeniobiorców,
10) istnienia alternatywnej technologii medycznej, w rozumieniu ustawy o świadczeniach, oraz jej efektywności klinicznej i bezpieczeństwa stosowania,
11) wiarygodności i precyzji oszacowań kryteriów, o których mowa w pkt 3–10,
12) priorytetów zdrowotnych określonych w przepisach wydanych na podstawie art. 31a ust. 2 ustawy o świadczeniach,
13) wysokości progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość, ustalonego w wysokości trzykrotności Produktu Krajowego Brutto na jednego mieszkańca, o którym mowa w art. 6 ust. 1 ustawy z dnia 26 października 2000 r. o sposobie obliczania wartości rocznego produktu krajowego brutto (Dz. U. poz. 1188 oraz z 2009 r. poz. 817), a w przypadku braku możliwości wyznaczenia tego kosztu – koszt uzyskania dodatkowego roku życia
– biorąc pod uwagę inne możliwe do zastosowania w danym stanie klinicznym procedury medyczne, które mogą być zastąpione przez wnioskowany lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny.
Art. 13. 1. Minister właściwy do spraw zdrowia ustala urzędową cenę zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, dla którego żaden odpowiednik nie jest refundowany w danym wskazaniu, przy uwzględnieniu następujących kryteriów:
1) stanowiska Komisji Ekonomicznej, o której mowa w art. 17,
2) rekomendacji Prezesa Agencji, o której mowa w art. 35 ust. 6, w szczególności wyników analizy stosunku kosztów do uzyskanych efektów zdrowotnych,
3) konkurencyjności cenowej
– biorąc pod uwagę równoważenie interesów świadczeniobiorców i przedsiębiorców zajmujących się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi, możliwości płatnicze podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych oraz działalność naukowo-badawczą i inwestycyjną wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA).
2. W pierwszej decyzji administracyjnej wydanej po zaistnieniu okoliczności, o których mowa w art. 11 ust. 3, urzędowa cena zbytu nie może być wyższa niż 75% urzędowej ceny zbytu określonej w poprzedniej decyzji administracyjnej o objęciu refundacją.
3. Jeżeli analiza kliniczna, o której mowa w art. 25 pkt 14 lit. c tiret pierwsze, nie zawiera randomizowanych badań klinicznych, dowodzących wyższości leku nad technologiami medycznymi, w rozumieniu ustawy o świadczeniach, dotychczas refundowanymi w danym wskazaniu, to urzędowa cena zbytu leku musi być skalkulowana w taki sposób, aby koszt stosowania leku wnioskowanego do objęcia refundacją nie był wyższy niż koszt technologii medycznej, w rozumieniu ustawy o świadczeniach, dotychczas finansowanej ze środków publicznych, o najkorzystniejszym współczynniku uzyskiwanych efektów zdrowotnych do kosztów ich uzyskania.
4. Urzędowa cena zbytu dla leku, w sytuacji, o której mowa w ust. 3, ustalona zostaje w decyzji administracyjnej o objęciu refundacją wyłącznie w ten sposób, aby koszt stosowania leku wnioskowanego do objęcia refundacją nie był wyższy niż koszt technologii medycznej, w rozumieniu ustawy o świadczeniach, dotychczas finansowanej ze środków publicznych, o najkorzystniejszym współczynniku uzyskiwanych efektów zdrowotnych do kosztów ich uzyskania.
5. Minister właściwy do spraw zdrowia ustala urzędową cenę zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, dla którego co najmniej jeden odpowiednik jest refundowany w danym wskazaniu, przy uwzględnieniu kryteriów, o których mowa w ust. 1, biorąc pod uwagę równoważenie interesów świadczeniobiorców i przedsiębiorców zajmujących się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego oraz wyrobami medycznymi, możliwości płatnicze podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych oraz działalność naukowo-badawczą i inwestycyjną wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA).
6. Urzędowa cena zbytu, o której mowa w ust. 5, z uwzględnieniem liczby DDD w opakowaniu jednostkowym, nie może być wyższa niż:
1) 75% urzędowej ceny zbytu jedynego odpowiednika refundowanego w danym wskazaniu;
2) urzędowa cena zbytu:
a) odpowiednika wyznaczającego podstawę limitu albo
b) najtańszego odpowiednika o ile podstawę limitu w danej grupie limitowej wyznacza lek z inną substancją czynną
– w przypadku kolejnego odpowiednika refundowanego w danym wskazaniu.
6a. Urzędowa cena zbytu ustalona w decyzji, o której mowa w art. 11 ust. 1, nie może być wyższa niż urzędowa cena zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, obowiązująca w dniu złożenia wniosku, o którym mowa w art. 24 ust. 1 pkt 1, w odniesieniu do leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który w dniu złożenia tego wniosku był zawarty w wykazie, o którym mowa w art. 37 ust. 1, w danym wskazaniu.
7. Przepis ust. 6 stosuje się odpowiednio do środka spożywczego specjalnego przeznaczenia żywieniowego i wyrobu medycznego.
8. Minister właściwy do spraw zdrowia ustala urzędową cenę zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, o którym mowa w art. 6 ust. 1 pkt 4, przy uwzględnieniu kryteriów:
1) stanowiska Komisji Ekonomicznej, o której mowa w art. 17,
2) minimalnej ceny zbytu netto, uzyskanej na terytorium Rzeczypospolitej Polskiej w okresie roku przed złożeniem wniosku dla wnioskowanej wielkości opakowania i dawki,
3) minimalnej ceny zbytu netto, uzyskanej w poszczególnych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA) w ramach finansowania ze środków publicznych tych państw, w okresie roku przed złożeniem wniosku, przeliczone w złotych polskich po średnim kursie Narodowego Banku Polskiego z miesiąca poprzedzającego miesiąc złożenia wniosku; w przypadku gdy przedmiot wniosku nie jest finansowany ze środków publicznych w danym państwie, uwzględnia się odpowiednio ceny uzyskane na wolnym rynku,
4) urzędowej ceny zbytu leków posiadających tę samą nazwę międzynarodową albo inne nazwy międzynarodowe, ale podobne działanie terapeutyczne oraz środków spożywczych specjalnego przeznaczenia żywieniowego, przy zastosowaniu następujących kryteriów:
a) tych samych wskazań lub przeznaczeń,
b) podobnej skuteczności
– biorąc pod uwagę równoważenie interesów świadczeniobiorców i przedsiębiorców zajmujących się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, możliwości płatnicze podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych oraz działalność naukowo-badawczą i inwestycyjną wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA).
Art. 14. 1. Minister właściwy do spraw zdrowia, wydając decyzję o objęciu refundacją, dokonuje kwalifikacji do następujących odpłatności:
1) bezpłatnie – leku, wyrobu medycznego mającego udowodnioną skuteczność w leczeniu nowotworu złośliwego, zaburzenia psychotycznego, upośledzenia umysłowego lub zaburzenia rozwojowego albo choroby zakaźnej o szczególnym zagrożeniu epidemicznym dla populacji, albo leku, środka spożywczego specjalnego przeznaczenia żywieniowego stosowanego w ramach programu lekowego;
2) ryczałtowej – leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego:
a) wymagającego, zgodnie z aktualną wiedzą medyczną, stosowania dłużej niż 30 dni oraz którego miesięczny koszt stosowania dla świadczeniobiorcy przy odpłatności 30% limitu finansowania przekraczałby 5% minimalnego wynagrodzenia za pracę, ogłaszanego w obwieszczeniu Prezesa Rady Ministrów wydanym na podstawie art. 2 ust. 4 ustawy z dnia 10 października 2002 r. o minimalnym wynagrodzeniu za pracę, albo
b) zakwalifikowanego na podstawie art. 72 lub jego odpowiednika, albo
c) wymagającego, zgodnie z aktualną wiedzą medyczną, stosowania nie dłużej niż 30 dni oraz którego koszt stosowania dla świadczeniobiorcy przy odpłatności 50% limitu finansowania przekraczałby 30% minimalnego wynagrodzenia za pracę, ogłaszanego w obwieszczeniu Prezesa Rady Ministrów wydanym na podstawie art. 2 ust. 4 ustawy z dnia 10 października 2002 r. o minimalnym wynagrodzeniu za pracę;
3) 50% – leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który wymaga, zgodnie z aktualną wiedzą medyczną, stosowania nie dłużej niż 30 dni;
4) 30% – leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który nie został zakwalifikowany do poziomów odpłatności określonych w pkt 1–3.
2. Kwalifikacji do odpowiedniej odpłatności dokonuje się przy założeniu stosowania jednej DDD dobowo. W przypadku braku DDD kwalifikacji dokonuje się w oparciu o koszt miesięcznej terapii.
Art. 15. 1. Minister właściwy do spraw zdrowia ustala grupy leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, w ramach których wyznacza się podstawę limitu. Grup limitowych nie tworzy się w odniesieniu do leków, środków spożywczych specjalnego przeznaczenia żywieniowego, o których mowa w art. 6 ust. 1 pkt 4.
2. Do grupy limitowej kwalifikuje się lek posiadający tę samą nazwę międzynarodową albo inne nazwy międzynarodowe, ale podobne działanie terapeutyczne i zbliżony mechanizm działania oraz środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny, przy zastosowaniu następujących kryteriów:
1) tych samych wskazań lub przeznaczeń, w których są refundowane;
2) podobnej skuteczności.
3. Po zasięgnięciu opinii Rady Przejrzystości, opierającej się w szczególności na porównaniu wielkości kosztów uzyskania podobnego efektu zdrowotnego lub dodatkowego efektu zdrowotnego, dopuszcza się tworzenie:
1) odrębnej grupy limitowej, w przypadku gdy droga podania leku lub jego postać farmaceutyczna w istotny sposób ma wpływ na efekt zdrowotny lub dodatkowy efekt zdrowotny;
2) wspólnej grupy limitowej, w przypadku gdy podobny efekt zdrowotny lub podobny dodatkowy efekt zdrowotny uzyskiwany jest pomimo odmiennych mechanizmów działania leków;
3) odrębnej grupy limitowej dla środków spożywczych specjalnego przeznaczenia żywieniowego, jeżeli zawartość składników odżywczych w istotny sposób wpływa na efekt zdrowotny lub dodatkowy efekt zdrowotny.
4. Podstawę limitu w danej grupie limitowej leków stanowi najwyższa spośród najniższych cen hurtowych za DDD leku, który dopełnia 15% obrotu ilościowego, liczonego według DDD, zrealizowanego w tej grupie limitowej w miesiącu poprzedzającym o 3 miesiące ogłoszenie obwieszczenia, o którym mowa w art. 37.
5. Podstawę limitu w przypadku:
1) środka spożywczego specjalnego przeznaczenia żywieniowego – stanowi najniższy koszt 30 dniowego stosowania według cen hurtowych;
2) wyrobu medycznego – stanowi najwyższa spośród najniższych cen hurtowych za jednostkę tego wyrobu medycznego, który dopełnia 15% obrotu ilościowego zrealizowanego w tej grupie limitowej w miesiącu poprzedzającym o 3 miesiące ogłoszenie obwieszczenia, o którym mowa w art. 37.
6. Jeżeli cena detaliczna jest niższa niż limit finansowania, limit finansowania ulega obniżeniu do wysokości ceny detalicznej tego leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego.
7. W przypadku wydania decyzji o objęciu refundacją pierwszego odpowiednika refundowanego w danym wskazaniu podstawą limitu w grupie limitowej jest cena hurtowa za DDD tego odpowiednika. W przypadku objęcia refundacją kolejnych odpowiedników podstawa limitu nie może być wyższa niż cena hurtowa za DDD pierwszego odpowiednika.
8. Jeżeli informacje o obrocie ilościowym, o którym mowa w ust. 4 i 5, nie są dostępne, wykorzystuje się informacje najbardziej aktualne.
9. Wysokość limitu finansowania za opakowanie jednostkowe jest równa iloczynowi kosztu DDD podstawy limitu i liczby DDD w opakowaniu jednostkowym, z uwzględnieniem urzędowej marży detalicznej. W przypadku gdy DDD nie jest określone do wyliczenia limitu finansowania przyjmuje się koszt terapii dziennej i ilość terapii dziennej w danym opakowaniu.
10. W przypadku środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego wysokość limitu finansowania jest równa iloczynowi kosztu jednostki podstawy limitu i ilości jednostek w danym opakowaniu.
11. Podstawę limitu w danej grupie limitowej leków, o których mowa w art. 6 ust. 1 pkt 2 i 3 stanowi najwyższa spośród najniższych cen hurtowych za DDD leku, który według deklaracji złożonej we wniosku o objęcie refundacją dopełnia 110% obrotu ilościowego, liczonego według DDD, zrealizowanego w tej grupie limitowej w roku poprzedzającym rok ustalenia podstawy albo 100% szacowanego zapotrzebowania w przypadku leku, dla którego zostanie utworzona nowa grupa limitowa.
12. Wysokość limitu finansowania leków, o których mowa w art. 6 ust. 1 pkt 2 i 3, świadczeniodawcom jest równa iloczynowi kosztu DDD podstawy limitu i liczby DDD podanych świadczeniobiorcom w ramach realizacji umowy o udzielanie świadczeń opieki zdrowotnej.
13. Przepisy ust. 11 i 12 stosuje się odpowiednio w przypadku:
1) leków, dla których DDD nie zostało określone,
2) środków spożywczych specjalnego przeznaczenia żywieniowego
– o których mowa w art. 6 ust. 1 pkt 2 i 3.
14. W przypadku, gdy DDD jest niższe od najczęściej stosowanej dobowo dawki leku (PDD) podstawę limitu minister właściwy do spraw zdrowia może wyznaczyć na podstawie PDD.
Art. 16. W przypadku, gdy w wyniku wydania decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, zachodzi konieczność utworzenia nowej grupy limitowej obejmującej również leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne zakwalifikowane uprzednio do innej grupy limitowej, minister właściwy do spraw zdrowia wydaje z urzędu decyzje administracyjne, zmieniające określenie grupy limitowej, o której mowa w art. 11 ust. 2 pkt 8.
Rozdział 4
Komisja Ekonomiczna
Art. 17. 1. Przy ministrze właściwym do spraw zdrowia tworzy się Komisję Ekonomiczną, zwaną dalej „Komisją”.
2. W skład Komisji wchodzi:
1) dwunastu przedstawicieli ministra właściwego do spraw zdrowia;
2) pięciu przedstawicieli Prezesa Funduszu.
3. Członkiem Komisji może zostać osoba:
1) która:
a) korzysta z pełni praw publicznych,
b) nie była skazana prawomocnym wyrokiem za umyślne przestępstwo lub umyślne przestępstwo skarbowe,
c) posiada wiedzę i doświadczenie, które dają rękojmię skutecznego prowadzenia negocjacji;
2) wobec której nie zachodzą okoliczności określone w art. 20 ust. 1 i art. 21.
4. Członków Komisji powołuje i odwołuje minister właściwy do spraw zdrowia.
Art. 18. 1. Do zadań Komisji należy prowadzenie negocjacji z wnioskodawcą w zakresie:
1) ustalenia urzędowej ceny zbytu;
2) poziomu odpłatności;
3) wskazań, w których lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny ma być refundowany;
4) instrumentów dzielenia ryzyka, o których mowa w art. 11 ust. 5.
2. Do zadań Komisji należy również:
1) monitorowanie realizacji całkowitego budżetu na refundację;
2) prowadzenie działań mających na celu racjonalizację wydatków związanych z refundacją oraz przedstawianie ministrowi właściwemu do spraw zdrowia propozycji w tym zakresie;
3) monitorowanie realizacji zobowiązania, o którym mowa w art. 25 pkt 4;
4) realizacja innych zadań zleconych przez ministra właściwego do spraw zdrowia.
3.6) Komisja na podstawie dokumentu stanowiącego wynik negocjacji, podpisanego przez strony negocjacji, przyjmuje stanowisko w drodze uchwały i przedstawia je niezwłocznie ministrowi właściwemu do spraw zdrowia.
3.7) Komisja na podstawie dokumentu stanowiącego wynik negocjacji, sporządzonego w postaci elektronicznej, podpisanego przez strony negocjacji, podejmuje uchwałę w drodze głosowania elektronicznego za pomocą Systemu Obsługi List Refundacyjnych, o którym mowa w art. 30a ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia (Dz. U. z 2015 r. poz. 636, z późn. zm.8)), zwanego dalej „SOLR”, i przekazuje ją niezwłocznie ministrowi właściwemu do spraw zdrowia.
Art. 19. 1.9) Rozpatrując wnioski, o których mowa w art. 24 ust. 1, Komisja prowadzi negocjacje w składzie pięcioosobowym, z tym że w każdym składzie powinien znaleźć się przedstawiciel Prezesa Funduszu.
1.10) Rozpatrując wnioski, o których mowa w art. 24 ust. 1, Komisja prowadzi negocjacje w składzie pięcioosobowym, zwanym dalej „zespołem negocjacyjnym”, z tym że w każdym składzie powinien znaleźć się przedstawiciel Prezesa Funduszu.
2. Negocjacje prowadzi się biorąc pod uwagę następujące kryteria:
1) rekomendację Prezesa Agencji, o której mowa w art. 35 ust. 6, w szczególności wyników analizy stosunku kosztów do uzyskanych efektów zdrowotnych,
2) maksymalną i minimalną cenę zbytu netto, uzyskaną na terytorium Rzeczypospolitej Polskiej w okresie roku przed złożeniem wniosku, o którym mowa w art. 24 ust. 1, dla danej wielkości opakowania i dawki, będącej przedmiotem tego wniosku, jeżeli dotyczy,
3) maksymalną i minimalną cenę zbytu netto, uzyskaną w poszczególnych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA), w ramach finansowania ze środków publicznych tych państw, w okresie roku przed złożeniem wniosku, o którym mowa w art. 24 ust. 1, przeliczoną w złotych polskich po średnim kursie Narodowego Banku Polskiego z miesiąca poprzedzającego miesiąc złożenia wniosku; w przypadku gdy przedmiot wniosku nie jest finansowany ze środków publicznych w danych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA), uwzględnia się odpowiednio ceny uzyskane na wolnym rynku,
4) informację o rabatach, upustach lub porozumieniach cenowych w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA),
5) koszt terapii przy zastosowaniu wnioskowanego leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego w porównaniu z innymi możliwymi do zastosowania w danym stanie klinicznym technologiami medycznymi, które mogą zostać zastąpione przez ten lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny,
6) wpływ na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych,
7) wysokość progu kosztu uzyskania dodatkowego roku życia skorygowanego o jego jakość, ustalonego w wysokości trzykrotności Produktu Krajowego Brutto na jednego mieszkańca, o którym mowa w art. 6 ust. 1 ustawy z dnia 26 października 2000 r. o sposobie obliczania wartości rocznego produktu krajowego brutto, a w przypadku braku możliwości wyznaczenia tego kosztu – koszt uzyskania dodatkowego roku życia
– uwzględniając potrzebę równoważenia interesów świadczeniobiorców i przedsiębiorców zajmujących się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi, możliwości płatnicze podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych oraz działalność naukowo-badawczą i inwestycyjną wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA).
3.11) Wynikiem negocjacji jest dokument sporządzony w postaci elektronicznej w trakcie negocjacji podpisany przez przewodniczącego zespołu negocjacyjnego oraz wnioskodawcę za pomocą podpisu potwierdzonego profilem zaufanym ePUAP w rozumieniu ustawy z dnia 17 lutego 2005 r. o informatyzacji działalności podmiotów realizujących zadania publiczne (Dz. U. z 2014 r. poz. 1114 oraz z 2016 r. poz. 352) lub bezpiecznym podpisem elektronicznym weryfikowanym przy pomocy ważnego kwalifikowanego certyfikatu w rozumieniu art. 3 pkt 12 ustawy z dnia 18 września 2001 r. o podpisie elektronicznym (Dz. U. z 2013 r. poz. 262, z 2014 r. poz. 1662 oraz z 2015 r. poz. 1893), przekazywany następnie Komisji w celu podjęcia uchwały.
Art. 20. 1. Członkowie Komisji, ich małżonkowie, zstępni i wstępni w linii prostej oraz osoby, z którymi członkowie Komisji pozostają we wspólnym pożyciu nie mogą:
1) być członkami organów spółek handlowych lub przedstawicielami przedsiębiorców prowadzących działalność gospodarczą w zakresie wytwarzania lub obrotu lekiem, środkiem spożywczym specjalnego przeznaczenia żywieniowego, wyrobem medycznym;
2) być członkami organów spółek handlowych lub przedstawicielami przedsiębiorców prowadzących działalność gospodarczą w zakresie doradztwa związanego z refundacją leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych;
3) być członkami organów spółdzielni, stowarzyszeń lub fundacji prowadzących działalność, o której mowa w pkt 1 i 2;
4) posiadać akcji lub udziałów w spółkach handlowych prowadzących działalność, o której mowa w pkt 1 i 2, oraz udziałów w spółdzielniach prowadzących działalność, o której mowa w pkt 1 i 2;
5) prowadzić działalności gospodarczej w zakresie, o którym mowa w pkt 1 i 2;
6) wykonywać zajęć zarobkowych na podstawie stosunku pracy, umowy o świadczenie usług zarządczych, umowy zlecenia, umowy o dzieło lub innej umowy o podobnym charakterze zawartej z podmiotami, o których mowa w pkt 1–3.
2. Członkowie Komisji składają ministrowi właściwemu do spraw zdrowia oświadczenie o niezachodzeniu okoliczności określonych w ust. 1, pod rygorem odpowiedzialności karnej za składanie fałszywych oświadczeń z art. 233 § 1 i 6 ustawy z dnia 6 czerwca 1997 r. – Kodeks karny (Dz. U. poz. 1137), zwane dalej „deklaracją o braku konfliktu interesów”, przed powołaniem oraz przed każdym posiedzeniem Komisji.
3. Zaistnienie okoliczności, o których mowa w ust. 1, stanowi podstawę do odwołania członka ze składu Komisji.
4. W przypadku zaistnienia okoliczności, o których mowa w ust. 1 pkt 4, niezależnych od członka Komisji, członek Komisji niezwłocznie informuje ministra właściwego do spraw zdrowia o tym fakcie. Minister właściwy do spraw zdrowia zawiesza członka w pracach Komisji, wyznaczając mu termin usunięcia zaistniałych okoliczności. Po bezskutecznym upływie tego terminu, minister właściwy do spraw zdrowia odwołuje członka Komisji.
5. Deklaracja o braku konfliktu interesów zawiera:
1) imię i nazwisko osoby składającej oświadczenie;
2) imię i nazwisko: małżonka, wstępnych, zstępnych w linii prostej oraz osób, z którymi członkowie Komisji pozostają we wspólnym pożyciu;
3) oświadczenie o niezachodzeniu okoliczności, o których mowa w ust. 1.
6. Deklaracje o braku konfliktu interesów weryfikuje Centralne Biuro Antykorupcyjne.
Art. 21. 1. Nie można być jednocześnie członkiem Komisji i Rady Przejrzystości.
2. Funkcji członka Komisji albo Rady Przejrzystości nie można łączyć z funkcją Prezesa Agencji i jego zastępcy.
Art. 22. 1. Działalność Komisji jest finansowana z budżetu państwa ze środków pozostających w dyspozycji ministra właściwego do spraw zdrowia.
2. Członkom Komisji przysługuje wynagrodzenie za udział w posiedzeniach w wysokości nieprzekraczającej 3500 zł za udział w każdym posiedzeniu Komisji, jednak nie więcej niż 10 500 zł miesięcznie.
3. Członkom Komisji przysługuje zwrot kosztów podróży na zasadach określonych w przepisach dotyczących zasad ustalania oraz wysokości należności przysługujących pracownikowi zatrudnionemu w państwowej lub samorządowej jednostce budżetowej z tytułu podróży służbowej na terenie kraju.
Art. 23. 1. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia:
1) wzór deklaracji o braku konfliktu interesów, mając na uwadze wymagania, o których mowa w art. 20 ust. 5;
2) wysokość wynagrodzenia członków Komisji, biorąc pod uwagę zakres działania Komisji oraz specyfikę wykonywanych zadań.
2. Minister właściwy do spraw zdrowia określi w drodze zarządzenia:
1) regulamin Komisji określający jej organizację, sposób i tryb działania, a także sposób wyłaniania i odwoływania przewodniczącego oraz członków Komisji;
2) tryb wyłaniania członków Komisji na poszczególne posiedzenia w sprawie prowadzenia negocjacji;
3) sposób obsługi organizacyjno-technicznej Komisji.
Rozdział 5
Tryb podejmowania decyzji w sprawie refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego i wyrobów medycznych
Art. 24. 1. Wnioskodawca może złożyć do ministra właściwego do spraw zdrowia wniosek o:
1) objęcie refundacją i ustalenie urzędowej ceny zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego;
2) podwyższenie urzędowej ceny zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego objętego refundacją;
3) obniżenie urzędowej ceny zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego objętego refundacją;
4) ustalenie albo zmianę urzędowej ceny zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, o którym mowa w art. 6 ust. 1 pkt 4;
5) skrócenie okresu obowiązywania decyzji, o której mowa w art. 11 ust. 1 albo ust. 6.
2. Do wniosków, o których mowa w ust. 1, dołącza się także:
1) informację aktualną na dzień złożenia wniosku dotyczącą refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego we wszystkich państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA) wraz z określeniem poziomu refundacji, jej warunków i ograniczeń, w tym szczegółowych informacji dotyczących zawartych instrumentów dzielenia ryzyka, albo informację o nieistnieniu takich ograniczeń lub niezawarciu takich instrumentów – informację tę potwierdza się właściwymi dokumentami przetłumaczonymi przysięgle na język polski;
2) aktualną na dzień złożenia wniosku: Charakterystykę Produktu Leczniczego albo oznakowanie środka spożywczego specjalnego przeznaczenia żywieniowego, albo instrukcję stosowania wyrobu medycznego, jeżeli dotyczy;
3) aktualny odpis z rejestru, do którego wnioskodawca jest wpisany, lub równoważny mu dokument wystawiony poza granicami Rzeczypospolitej Polskiej, wydany nie wcześniej niż 3 miesiące przed dniem złożenia wniosku; w przypadku wnioskodawców zagranicznych należy dodatkowo dołączyć tłumaczenie przysięgłe odpowiedniego dokumentu na język polski;
4) upoważnienie do reprezentowania wnioskodawcy, jeżeli dotyczy;
5) umowę zawartą pomiędzy podmiotem odpowiedzialnym a przedstawicielem podmiotu odpowiedzialnego, w rozumieniu przepisów ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, jeżeli dotyczy.
3.12) Wnioskodawca składa odrębny wniosek dla każdej dawki i wielkości opakowania leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego.
3.13) Wnioskodawca składa odrębny wniosek dla każdej dawki, wielkości opakowania, kategorii dostępności refundacyjnej, o której mowa w art. 6 ust. 1, lub poziomu odpłatności leku, środka spożywczego specjalnego przeznaczenia żywieniowego oraz wyrobu medycznego.
4. W przypadku gdy analizy, o których mowa w art. 26 pkt 1 lit. h oraz i lub pkt 2 lit. h–j, są właściwe dla więcej niż jednego wniosku, dopuszcza się złożenie tych analiz jako wspólnych załączników do składanych wniosków.
5. W przypadku wnioskowania o objęcie refundacją dodatkowego wskazania dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo dodatkowego zastosowania wyrobu medycznego wnioskodawca składa wniosek, o którym mowa w ust. 1 pkt 1.
6.14) Wnioski wraz z załącznikami składa się w formie pisemnej i elektronicznej.
6.15) Wnioski wraz z załącznikami oraz inne wnioski, pisma ministra właściwego do spraw zdrowia oraz pisma strony składane w postępowaniu w zakresie wydania decyzji administracyjnej w sprawie refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego składa się w postaci elektronicznej za pomocą SOLR.
6a.16) Wnioski, o których mowa w ust. 1, oraz dokumenty, o których mowa w ust. 2, opatruje się podpisem potwierdzonym profilem zaufanym ePUAP w rozumieniu ustawy z dnia 17 lutego 2005 r. o informatyzacji działalności podmiotów realizujących zadania publiczne lub bezpiecznym podpisem elektronicznym weryfikowanym przy pomocy ważnego kwalifikowanego certyfikatu w rozumieniu art. 3 pkt 12 ustawy z dnia 18 września 2001 r. o podpisie elektronicznym.
6b.16) Z wnioskodawcami, o których mowa w ust. 1, minister właściwy do spraw zdrowia komunikuje się za pomocą SOLR.
6c.16) W przypadku braku dostępu do SOLR na skutek awarii systemu lub działania siły wyższej, termin na dokonanie czynności dotyczących postępowania w sprawie refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego uznaje się za zachowany pod warunkiem dokonania w wyznaczonym terminie czynności w postaci papierowej. Dokumentacja złożona w postaci papierowej, po uzyskaniu dostępu do SOLR, jest wprowadzana przez ministra właściwego do spraw zdrowia do systemu w terminie trzech dni roboczych od dnia przywrócenia funkcjonalności systemu.
6d.16) W uzasadnionych przypadkach, gdy wystąpi brak dostępu do SOLR na skutek awarii systemu lub działania siły wyższej, jest możliwe dokonanie czynności w zakresie wydania decyzji administracyjnej w sprawie refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego bez pośrednictwa SOLR. Przepis ust. 6c w zakresie wprowadzania do systemu dokumentacji stosuje się odpowiednio.
7. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia:
1)17) wzór wniosku, o którym mowa w ust. 1, sposób jego składania oraz format przekazywanych danych, mając na względzie zapewnienie jednolitości zakresu i rodzaju danych, a także bezpieczeństwa przekazywania tych danych;
1) (uchylony)18)
2) minimalne wymagania, jakie muszą spełniać analizy, o których mowa w art. 25 pkt 14 lit. c i art. 26 pkt 2 lit. h–j, biorąc pod uwagę potrzebę zapewnienia niezbędnej wiarygodności i precyzji tych analiz, koniecznej do podjęcia na ich podstawie adekwatnych decyzji o objęciu refundacją.
Art. 25. Wniosek, o którym mowa w art. 24 ust. 1 pkt 1, zawiera:
1) oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej, imię i nazwisko, telefon, telefaks, adres poczty elektronicznej i adres korespondencyjny osoby upoważnionej do jego reprezentowania w sprawie tego wniosku;
2) określenie przedmiotu wniosku;
3) dowód dostępności w obrocie leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, w chwili składania wniosku;
4)19) zobowiązanie do zapewnienia ciągłości dostaw wraz z określeniem rocznej wielkości dostaw podanej w ujęciu miesięcznym, w przypadku objęcia refundacją;
5) dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny w danej wielkości i dawce, jeżeli dotyczy:
a)20) nazwę, jego postać, drogę podania albo sposób zastosowania oraz rodzaj opakowania,
a)21) nazwę, jego postać, rodzaj, drogę podania albo sposób zastosowania oraz rodzaj opakowania,
b)20) numer pozwolenia oraz kopię decyzji o dopuszczeniu do obrotu,
b)21) numer pozwolenia oraz kopię decyzji o dopuszczeniu do obrotu leku albo kopię powiadomienia o wprowadzeniu do obrotu środka spożywczego specjalnego przeznaczenia żywieniowego albo kopię powiadomienia lub zgłoszenia wyrobu medycznego,
c) kod identyfikacyjny EAN lub inny kod odpowiadający kodowi EAN;
6) wnioskowane warunki objęcia refundacją, w szczególności:
a) wskazania, w których lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny ma być refundowany,
b) proponowaną cenę zbytu netto,
c) kategorię dostępności refundacyjnej, o której mowa w art. 6 ust. 1,
d) poziom odpłatności,
e) instrumenty dzielenia ryzyka, o których mowa w art. 11 ust. 5,
f) okres obowiązywania decyzji o objęciu refundacją,
g) projekt opisu programu lekowego, jeżeli dotyczy, zawierający:
– nazwę programu,
– cel programu,
– opis problemu medycznego,
– opis programu obejmujący: kryteria włączenia do programu, dawkowanie i sposób podawania, monitorowanie programu, w tym monitorowanie leczenia i sposób przekazywania informacji sprawozdawczo-rozliczeniowych, a także kryteria wyłączenia z programu;
7) wskazanie maksymalnej i minimalnej ceny zbytu netto, uzyskanej na terytorium Rzeczypospolitej Polskiej w okresie roku przed złożeniem wniosku dla wnioskowanej wielkości opakowania i dawki;
8) wskazanie maksymalnej i minimalnej ceny zbytu netto, uzyskanej w poszczególnych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA) w ramach finansowania ze środków publicznych tych państw w okresie roku przed złożeniem wniosku, przeliczone w złotych polskich po średnim kursie Narodowego Banku Polskiego z miesiąca poprzedzającego miesiąc złożenia wniosku; w przypadku gdy przedmiot wniosku nie jest finansowany ze środków publicznych w danym państwie, uwzględnia się odpowiednio ceny uzyskane na wolnym rynku; w przypadku wnioskodawcy będącego importerem równoległym wskazanie ceny zbytu netto leku z państwa, z którego jest sprowadzany;
9)22) dzienny koszt terapii, odrębnie dla każdego wskazania określonego w pkt 6 lit. a;
9)23) dzienny koszt terapii dla leku, odrębnie dla każdego wskazania określonego w pkt 6 lit. a;
10)22) średni koszt standardowej terapii, odrębnie dla każdego wskazania określonego w pkt 6 lit. a;
10)23) średni koszt standardowej terapii dla leku, odrębnie dla każdego wskazania określonego w pkt 6 lit. a;
11)22) czas trwania standardowej terapii, odrębnie dla każdego wskazania określonego w pkt 6 lit. a;
11)23) czas trwania standardowej terapii dla leku oraz środka spożywczego specjalnego przeznaczenia żywieniowego, odrębnie dla każdego wskazania określonego w pkt 6 lit. a;
12) informacje dotyczące terminu wygaśnięcia ochrony patentowej, w tym także dodatkowego świadectwa ochronnego, jeżeli dotyczy;
13)24) informacje dotyczące upływu okresu wyłączności danych oraz wyłączności rynkowej;
13)25) informacje dotyczące upływu okresu wyłączności danych oraz wyłączności rynkowej, jeżeli dotyczy;
14) uzasadnienie wniosku zawierające:
a) dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który ma co najmniej jeden odpowiednik refundowany w danym wskazaniu – analizę wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych,
b)26) w przypadku, o którym mowa w art. 40, co najmniej jedną publikację naukową potwierdzającą zasadność wniosku,
b) (uchylona)27)
c) dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który nie ma odpowiednika refundowanego w danym wskazaniu:
– analizę kliniczną, sporządzoną na podstawie przeglądu systematycznego w porównaniu z innymi możliwymi do zastosowania w danym stanie klinicznym procedurami medycznymi we wnioskowanym wskazaniu, w tym, o ile występują, finansowanymi ze środków publicznych,
– analizę ekonomiczną z perspektywy podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych oraz świadczeniobiorcy,
– analizę wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych,
– analizę racjonalizacyjną, przedkładaną w przypadku gdy analiza wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych wykazuje wzrost kosztów refundacji; analiza ta powinna przedstawiać rozwiązania dotyczące refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, których objęcie refundacją spowoduje uwolnienie środków publicznych w wielkości odpowiadającej co najmniej wzrostowi kosztów wynikającemu z analizy wpływu na budżet,
d) informacje dotyczące działalności naukowo-badawczej i inwestycyjnej wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA);
15) dowód uiszczenia opłaty, o której mowa w art. 32 i art. 35 ust. 3, jeżeli dotyczy.
Art. 26. Wniosek, o którym mowa w art. 24 ust. 1 pkt 2, zawiera, jeżeli dany lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny:
1) ma odpowiednik refundowany w danym wskazaniu:
a) oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej, imię i nazwisko, telefon, telefaks, adres poczty elektronicznej i adres korespondencyjny osoby upoważnionej do jego reprezentowania w sprawie tego wniosku,
b) określenie przedmiotu wniosku,
ba)28) numer decyzji, której urzędowa cena zbytu ma ulec zmianie,
c) proponowaną cenę zbytu netto,
d) dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny w danej wielkości i dawce, jeżeli dotyczy:
–29) nazwę, jego postać, drogę podania albo sposób zastosowania oraz rodzaj opakowania,
–30) nazwę, jego postać, rodzaj, drogę podania albo sposób zastosowania oraz rodzaj opakowania,
–29) numer pozwolenia oraz kopię decyzji dopuszczenia do obrotu,
–30) numer pozwolenia oraz kopię decyzji o dopuszczeniu do obrotu leku albo kopię powiadomienia o wprowadzeniu do obrotu środka spożywczego specjalnego przeznaczenia żywieniowego albo kopię powiadomienia lub zgłoszenia wyrobu medycznego,
– kod identyfikacyjny EAN lub inny kod odpowiadający kodowi EAN,
e) wskazanie minimalnej ceny zbytu netto, uzyskanej na terytorium Rzeczypospolitej Polskiej w okresie roku przed złożeniem wniosku dla wnioskowanej wielkości opakowania i dawki,
f) wskazanie maksymalnej i minimalnej ceny zbytu netto, uzyskanej w poszczególnych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA) w ramach finansowania ze środków publicznych tych państw w okresie roku przed złożeniem wniosku, przeliczone w złotych polskich po średnim kursie Narodowego Banku Polskiego z miesiąca poprzedzającego miesiąc złożenia wniosku; w przypadku gdy przedmiot wniosku nie jest finansowany ze środków publicznych w danym państwie, uwzględnia się odpowiednio ceny uzyskane na wolnym rynku; w przypadku wnioskodawcy będącego importerem równoległym wskazanie ceny zbytu netto leku z państwa, z którego jest sprowadzany,
g) informacje dotyczące działalności naukowo-badawczej i inwestycyjnej wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA),
h) analizę wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych,
i) analizę racjonalizacyjną przedkładaną w przypadku gdy analiza wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych wykazuje wzrost kosztów refundacji; analiza ta powinna przedstawiać rozwiązania dotyczące refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, których wprowadzenie spowoduje uwolnienie środków publicznych w wielkości odpowiadającej co najmniej wzrostowi kosztów wynikającemu z analizy wpływu na budżet,
j)31) dzienny koszt terapii odrębnie dla każdego wskazania objętego refundacją,
j)32) dzienny koszt terapii dla leku, odrębnie dla każdego wskazania objętego refundacją,
k)31) średni koszt standardowej terapii, odrębnie dla każdego wskazania objętego refundacją,
k)32) średni koszt standardowej terapii dla leku, odrębnie dla każdego wskazania objętego refundacją,
l)31) czas trwania standardowej terapii, odrębnie dla każdego wskazania objętego refundacją,
l)32) czas trwania standardowej terapii dla leku oraz środka spożywczego specjalnego przeznaczenia żywieniowego, odrębnie dla każdego wskazania objętego refundacją,
m) dowód uiszczenia opłaty, o której mowa w art. 32 i art. 35 ust. 3, jeżeli dotyczy;
2) nie ma odpowiednika refundowanego w danym wskazaniu:
a) oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej, imię i nazwisko, telefon, telefaks, adres poczty elektronicznej i adres korespondencyjny osoby upoważnionej do jego reprezentowania w sprawie tego wniosku,
b) określenie przedmiotu wniosku,
ba)33) numer decyzji, której urzędowa cena zbytu ma ulec zmianie,
c) proponowaną cenę zbytu netto,
d) dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny w danej wielkości i dawce, jeżeli dotyczy:
–34) nazwę, jego postać, drogę podania albo sposób zastosowania oraz rodzaj opakowania,
–35) nazwę, jego postać, rodzaj, drogę podania albo sposób zastosowania oraz rodzaj opakowania,
–34) numer pozwolenia oraz kopię decyzji o dopuszczeniu do obrotu,
–35) numer pozwolenia oraz kopię decyzji o dopuszczeniu do obrotu leku albo kopię powiadomienia o wprowadzeniu do obrotu środka spożywczego specjalnego przeznaczenia żywieniowego albo kopię powiadomienia lub zgłoszenia wyrobu medycznego,
– kod identyfikacyjny EAN lub inny kod odpowiadający kodowi EAN,
e) wskazanie minimalnej ceny zbytu netto, uzyskanej na terytorium Rzeczypospolitej Polskiej w okresie roku przed złożeniem wniosku dla wnioskowanej wielkości opakowania i dawki,
f) wskazanie maksymalnej i minimalnej ceny zbytu netto, uzyskanej w poszczególnych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA), w ramach finansowania ze środków publicznych tych państw w okresie roku przed złożeniem wniosku, przeliczone w złotych polskich po średnim kursie Narodowego Banku Polskiego z miesiąca poprzedzającego miesiąc złożenia wniosku; w przypadku gdy przedmiot wniosku nie jest finansowany ze środków publicznych w danym państwie, uwzględnia się odpowiednio ceny uzyskane na wolnym rynku; w przypadku wnioskodawcy będącego importerem równoległym wskazanie ceny zbytu netto leku z państwa, z którego jest sprowadzany,
g) informacje dotyczące działalności naukowo-badawczej i inwestycyjnej wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA),
h) analizę kliniczną i ekonomiczną, jeżeli w uzasadnieniu wniosku są podane argumenty związane z efektem zdrowotnym, dodatkowym efektem zdrowotnym lub kosztami ich uzyskania,
i) analizę wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych,
j) analizę racjonalizacyjną przedkładaną w przypadku gdy analiza wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych wykazuje wzrost kosztów refundacji; analiza ta powinna przedstawiać rozwiązania dotyczące refundacji leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, których wprowadzenie spowoduje uwolnienie środków publicznych w wielkości odpowiadającej co najmniej wzrostowi kosztów wynikającemu z analizy wpływu na budżet,
k)36) dzienny koszt terapii, odrębnie dla każdego wskazania objętego refundacją,
k)37) dzienny koszt terapii dla leku, odrębnie dla każdego wskazania objętego refundacją,
l)36) średni koszt standardowej terapii, odrębnie dla każdego wskazania objętego refundacją,
l)37) średni koszt standardowej terapii dla leku, odrębnie dla każdego wskazania objętego refundacją,
m)36) czas trwania standardowej terapii odrębnie dla każdego wskazania objętego refundacją,
m)37) czas trwania standardowej terapii dla leku oraz środka spożywczego specjalnego przeznaczenia żywieniowego, odrębnie dla każdego wskazania objętego refundacją,
n) dowód uiszczenia opłaty, o której mowa w art. 32 i art. 35 ust. 3, jeżeli dotyczy.
Art. 27. Wniosek, o którym mowa w art. 24 ust. 1 pkt 3, zawiera:
1) oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej, imię i nazwisko, telefon, telefaks, adres poczty elektronicznej i adres korespondencyjny osoby upoważnionej do jego reprezentowania w sprawie tego wniosku;
2) określenie przedmiotu wniosku;
3) proponowaną cenę zbytu netto;
4) dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny w danej wielkości i dawce, jeżeli dotyczy:
a)38) nazwę, jego postać, drogę podania albo sposób zastosowania oraz rodzaj opakowania,
a)39) nazwę, jego postać, rodzaj, drogę podania albo sposób zastosowania oraz rodzaj opakowania,
b)38) numer pozwolenia oraz kopię decyzji dopuszczenia do obrotu,
b)39) numer pozwolenia oraz kopię decyzji o dopuszczeniu do obrotu leku albo kopię powiadomienia o wprowadzeniu do obrotu środka spożywczego specjalnego przeznaczenia żywieniowego, albo kopię powiadomienia lub zgłoszenia wyrobu medycznego,
ba)40) numer decyzji, której urzędowa cena zbytu ma ulec zmianie,
c) kod identyfikacyjny EAN lub inny kod odpowiadający kodowi EAN;
5)41) dzienny koszt terapii, odrębnie dla każdego wskazania objętego refundacją;
5) (uchylony)42)
6)41) średni koszt standardowej terapii, odrębnie dla każdego wskazania objętego refundacją;
6) (uchylony)42)
7)41) czas trwania standardowej terapii, odrębnie dla każdego wskazania objętego refundacją.
7) (uchylony)42)
Art. 28. Wniosek, o którym mowa w art. 24 ust. 1 pkt 4, zawiera:
1) oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej, imię i nazwisko, telefon, telefaks, adres poczty elektronicznej i adres korespondencyjny osoby upoważnionej do jego reprezentowania w sprawie tego wniosku;
2) określenie przedmiotu wniosku;
3) proponowaną cenę zbytu netto;
4) dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego w danej wielkości i dawce, jeżeli dotyczy:
a) nazwę, jego postać, drogę podania oraz rodzaj opakowania,
b) numer pozwolenia oraz kopię decyzji o dopuszczeniu do obrotu,
c) kod identyfikacyjny EAN lub inny kod odpowiadający kodowi EAN;
5) wskazanie maksymalnej i minimalnej ceny zbytu netto, uzyskanej na terytorium Rzeczypospolitej Polskiej w okresie roku przed złożeniem wniosku dla wnioskowanej wielkości opakowania i dawki;
6) wskazanie maksymalnej i minimalnej ceny zbytu netto, uzyskanej w poszczególnych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA) w ramach finansowania ze środków publicznych tych państw w okresie roku przed złożeniem wniosku, przeliczone w złotych polskich po średnim kursie Narodowego Banku Polskiego z miesiąca poprzedzającego miesiąc złożenia wniosku; w przypadku gdy przedmiot wniosku nie jest finansowany ze środków publicznych w danym państwie, uwzględnia się odpowiednio ceny uzyskane na wolnym rynku; w przypadku wnioskodawcy będącego importerem równoległym wskazanie ceny zbytu netto leku z państwa, z którego jest sprowadzany;
7) uzasadnienie wniosku zawierające:
a) analizę wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych,
b) analizę racjonalizacyjną przedkładaną w przypadku gdy analiza wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych wykazuje wzrost kosztów refundacji; analiza ta powinna przedstawiać rozwiązania dotyczące refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych, których objęcie refundacją spowoduje uwolnienie środków publicznych w wielkości odpowiadającej co najmniej wzrostowi kosztów wynikającemu z analizy wpływu na budżet,
c) informacje dotyczące działalności naukowo-badawczej i inwestycyjnej wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA);
8) dowód uiszczenia opłaty, o której mowa w art. 32, jeżeli dotyczy.
Art. 29. Wniosek, o którym mowa w art. 24 ust. 1 pkt 5, zawiera:
1) oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej, imię i nazwisko, telefon, telefaks, adres poczty elektronicznej i adres korespondencyjny osoby upoważnionej do jego reprezentowania w sprawie tego wniosku;
2) określenie przedmiotu wniosku;
3) numer decyzji ulegającej skróceniu;
4) uzasadnienie wniosku;
5) analizę wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych;
6) analizę wpływu na wysokość odpłatności i dopłat świadczeniobiorców.
Art. 30. 1. Wymagania, o których mowa w art. 24 ust. 2 pkt 1, art. 25 pkt 12 i 13, art. 26 pkt 1 lit. g oraz pkt 2 lit. g, art. 28 pkt 7 lit. c, nie dotyczą wniosków, o których mowa w art. 24 ust. 1, składanych przez wnioskodawcę będącego importerem równoległym.
2. Wymagania, o których mowa w art. 25 pkt 14, nie dotyczą wniosków, o których mowa w art. 24 ust. 1 pkt 1, w odniesieniu do leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który w dniu złożenia tego wniosku był zawarty w wykazie, o którym mowa w art. 37 ust. 1, w danym wskazaniu. Do wniosków tych nie stosuje się przepisów art. 12 pkt 2, art. 13 ust. 1 pkt 2 i ust. 3 oraz art. 35.
Art. 31. 1. Wnioski, o których mowa w art. 24 ust. 1, są rozpatrywane według kolejności ich wpływu.
2. W pierwszej kolejności, rozpatrywane są wnioski, o których mowa w art. 24 ust. 1 pkt 3.
3. W przypadku, gdy wniosek, o którym mowa w art. 24 ust. 1, nie zawiera wymaganych danych, minister właściwy do spraw zdrowia niezwłocznie informuje wnioskodawcę o konieczności jego uzupełnienia, zgodnie z przepisami ustawy z dnia 14 czerwca 1960 r. – Kodeks postępowania administracyjnego (Dz. U. z 2016 r. poz. 23, 868 i 996).
4. Wniosek, o którym mowa w art. 24 ust. 1 pkt 1, albo wniosek o ponowne rozpatrzenie sprawy dotyczący tego wniosku, rozpatruje się w terminie 180 dni, z tym że w przypadku konieczności uzupełnienia danych niezbędnych do rozpatrzenia wniosku, bieg tego terminu ulega zawieszeniu do dnia otrzymania uzupełnienia danych albo do dnia upływu terminu uzupełnienia wniosku.
5. Wniosek, o którym mowa w art. 24 ust. 1 pkt 2, albo wniosek o ponowne rozpatrzenie sprawy dotyczący tego wniosku, rozpatruje się w terminie 90 dni, z tym że w przypadku konieczności uzupełnienia danych niezbędnych do rozpatrzenia wniosku, bieg tego terminu ulega zawieszeniu do dnia otrzymania uzupełnienia danych albo do dnia upływu terminu uzupełnienia wniosku.
6. Wniosek, o którym mowa w art. 24 ust. 1 pkt 4, albo wniosek o ponowne rozpatrzenie sprawy dotyczący tego wniosku, rozpatruje się w terminie 90 dni, z tym że w przypadku konieczności uzupełnienia danych niezbędnych do rozpatrzenia wniosku, bieg tego terminu ulega zawieszeniu do dnia otrzymania uzupełnienia danych albo do dnia upływu terminu uzupełnienia wniosku.
7. Wniosek, o którym mowa w art. 24 ust. 1 pkt 3, albo wniosek o ponowne rozpatrzenie sprawy dotyczący tego wniosku, rozpatruje się w terminie 30 dni, z tym że w przypadku konieczności uzupełnienia danych niezbędnych do rozpatrzenia wniosku, bieg tego terminu ulega zawieszeniu do dnia otrzymania uzupełnienia danych albo do dnia upływu terminu uzupełnienia wniosku.
8. W przypadku złożenia wniosków, o których mowa w art. 24 ust. 1 pkt 2, w liczbie przekraczającej o więcej niż 10% przeciętną liczbę wniosków, termin ich rozpatrzenia może być jednorazowo przedłużony o 60 dni. W takim przypadku minister właściwy do spraw zdrowia niezwłocznie informuje wnioskodawcę o przedłużeniu terminu, o którym mowa w ust. 5. Podstawą ustalenia przeciętnej liczby wniosków jest średnia liczba wniosków będących podstawą ustalenia trzech poprzednich wykazów, o których mowa w art. 37.
9. Jeżeli wniosek, o którym mowa w art. 24 ust. 1 pkt 2 albo 3, nie zostanie rozpatrzony w terminie, o którym mowa odpowiednio w ust. 5 albo 7 z uwzględnieniem ust. 8, to w decyzji ustala się cenę określoną we wniosku.
10. W przypadku wniosku, o którym mowa w art. 24 ust. 1 pkt 1, w zakresie dotyczącym ustalenia kategorii dostępności refundacyjnej, o której mowa w art. 6 ust. 1 pkt 2, termin, o którym mowa w ust. 4, ulega zawieszeniu do czasu uzgodnienia treści programu lekowego pomiędzy wnioskodawcą a ministrem właściwym do spraw zdrowia.
11. Uzgadnianie treści programu lekowego nie może trwać dłużej niż 60 dni. W przypadku nieuzgodnienia treści programu lekowego, minister właściwy do spraw zdrowia wydaje decyzję administracyjną o odmowie objęcia refundacją.
12. Wnioskodawca może przed złożeniem wniosku zwrócić się do ministra właściwego do spraw zdrowia o wydanie wstępnej opinii dotyczącej projektu programu lekowego. Do wydania wstępnej opinii nie stosuje się przepisów ustawy z dnia 14 czerwca 1960 r. – Kodeks postępowania administracyjnego.
Art. 32. 1. Za złożenie wniosku, o którym mowa w art. 24 ust. 1 pkt 1, 2, 4 i 5, oraz za jego uzupełnienie, o którym mowa w art. 31 ust. 3, pobiera się opłaty wnoszone na rachunek urzędu obsługującego ministra właściwego do spraw zdrowia.
2. Opłaty, o których mowa w ust. 1, stanowią dochód budżetu państwa i każda z nich nie może być wyższa niż 10 000 zł.
3. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, wysokość opłat, o których mowa w ust. 1, mając na uwadze nakład pracy i poziom kosztów związanych z rozpatrywaniem wniosków, o których mowa w art. 24 ust. 1.
Art. 32a.43) 1. Wnioskodawca składa do ministra właściwego do spraw zdrowia wniosek o założenie konta w SOLR umożliwiającego za pomocą tego systemu:
1) składanie wniosków w postaci elektronicznej;
2) komunikowanie się z ministrem właściwym do spraw zdrowia.
2. Wniosek o założenie konta w SOLR zawiera oznaczenie (firmę) wnioskodawcy, adres siedziby albo miejsca wykonywania działalności gospodarczej, a także imię i nazwisko, telefon, telefaks, adres poczty elektronicznej i adres korespondencyjny osoby upoważnionej do jego reprezentowania w sprawie tego wniosku.
3. Do wniosku o założenie konta w SOLR dołącza się:
1) aktualny odpis z rejestru, do którego wnioskujący o założenie konta jest wpisany, lub równoważny mu dokument wystawiony poza granicami Rzeczypospolitej Polskiej, wydany nie wcześniej niż 3 miesiące przed dniem złożenia wniosku; w przypadku wnioskodawców zagranicznych należy dodatkowo dołączyć tłumaczenie odpowiedniego dokumentu na język polski sporządzone i poświadczone przez tłumacza przysięgłego albo sprawdzone i poświadczone przez tłumacza przysięgłego, wykonującego zawód tłumacza przysięgłego na warunkach określonych w ustawie z dnia 25 listopada 2004 r. o zawodzie tłumacza przysięgłego (Dz. U. z 2015 r. poz. 487 i 1505 oraz z 2016 r. poz. 65), lub przez tłumacza przysięgłego mającego siedzibę na terytorium państwa członkowskiego;
2) upoważnienie do reprezentowania wnioskodawcy, jeżeli dotyczy;
3) dowód uiszczenia opłaty za założenie konta.
4. Wniosek o założenie konta w SOLR wraz z załącznikami składa się w postaci papierowej i elektronicznej.
5. Opłatę za złożenie wniosku w wysokości 500 zł wnosi się na rachunek bankowy urzędu obsługującego ministra właściwego do spraw zdrowia.
6. Opłata za złożenie wniosku stanowi dochód budżetu państwa.
7. Po rozpatrzeniu wniosku o założenie konta w SOLR, minister właściwy do spraw zdrowia zakłada wnioskodawcy konto (login i hasło), które służy do administrowania udostępnioną wnioskodawcy częścią SOLR. Login i hasło przesyła się w terminie 21 dni od dnia otrzymania wniosku na wskazany we wniosku adres poczty elektronicznej.
8. Wniosek o założenie konta w SOLR złożony w postaci elektronicznej opatruje się podpisem potwierdzonym profilem zaufanym ePUAP w rozumieniu ustawy z dnia 17 lutego 2005 r. o informatyzacji działalności podmiotów realizujących zadania publiczne lub bezpiecznym podpisem elektronicznym weryfikowanym przy pomocy ważnego kwalifikowanego certyfikatu w rozumieniu ustawy z dnia 18 września 2001 r. o podpisie elektronicznym.
Art. 33. 1. Minister właściwy do spraw zdrowia uchyla decyzję administracyjną o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, w przypadku:
1) stwierdzenia braku deklarowanej skuteczności terapeutycznej;
2) stwierdzenia ryzyka stosowania niewspółmiernego do efektu terapeutycznego;
3) podważenia wiarygodności i precyzji oszacowań kryteriów, o których mowa w art. 12 pkt 3–10;
4) gdy zobowiązanie, o którym mowa w art. 25 pkt 4, nie zostanie dotrzymane w zakresie dotyczącym zapewnienia ciągłości dostaw lub rocznej wielkości dostaw, i nastąpi niezaspokojenie potrzeb świadczeniobiorców.
2. W przypadkach, o których mowa w ust. 1 pkt 1–3, minister właściwy do spraw zdrowia uchyla decyzję administracyjną o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego po zasięgnięciu opinii Rady Przejrzystości.
3. Ostateczna decyzja uchylająca decyzję administracyjną o objęciu refundacją stanowi podstawę aktualizacji wykazów, o których mowa w art. 37.
Art. 34. 1. W przypadku gdy zobowiązanie, o którym mowa w art. 25 pkt 4, w zakresie dotyczącym rocznej wielkości dostaw lub ciągłości dostaw, nie zostanie dotrzymane i nastąpi niezaspokojenie potrzeb świadczeniobiorców, wnioskodawca, który otrzymał decyzję administracyjną o objęciu refundacją jest obowiązany do zwrotu do Funduszu kwoty stanowiącej iloczyn liczby niedostarczonych jednostkowych opakowań leku, środka spożywczego specjalnego przeznaczenia żywieniowego albo jednostkowych wyrobów medycznych i ich urzędowej ceny zbytu netto, chyba że niewykonywanie tego zobowiązania jest następstwem działania siły wyższej albo potrzeby świadczeniobiorców zostały zaspokojone przez jego odpowiednik.
2. Przez niedotrzymanie zobowiązania dotyczącego ciągłości dostaw, o którym mowa w ust. 1, rozumie się brak obrotu produktem objętym refundacją stwierdzony na podstawie raportów, o których mowa w art. 78 ust. 1 pkt 6 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne lub informacji, o których mowa w art. 190 ust. 2 ustawy o świadczeniach.
3. Przez niedotrzymanie zobowiązania dotyczącego wielkości rocznych dostaw, o którym mowa w ust 1, rozumie się niewprowadzenie w ciągu roku kalendarzowego do obrotu zadeklarowanej we wniosku o objęcie refundacją ilości leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego.
4. Kwotę, o której mowa w ust. 1, oblicza Fundusz na podstawie danych, o których mowa w art. 102 ust. 5 pkt 31 ustawy o świadczeniach.
5. Zestawienie kwot, o których mowa w ust. 4, Prezes Funduszu niezwłocznie przekazuje ministrowi właściwemu do spraw zdrowia.
6. Kwotę, o której mowa w ust. 1, ustala w drodze decyzji administracyjnej minister właściwy do spraw zdrowia i podlega ona uiszczeniu w terminie 30 dni od dnia, w którym decyzja stała się ostateczna.
Art. 35. 1. Kopię wniosku, o którym mowa w art. 24 ust. 1 pkt 1, dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który nie ma odpowiednika refundowanego w danym wskazaniu, wraz z analizami, o których mowa w art. 25 pkt 14 lit. c, a także kopię wniosku, o którym mowa w art. 24 ust. 1 pkt 2, dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który nie ma odpowiednika refundowanego w danym wskazaniu, jeżeli w uzasadnieniu wniosku są podane argumenty związane z efektem zdrowotnym, dodatkowym efektem zdrowotnym lub kosztami ich uzyskania wraz z analizami, o których mowa w art. 26 pkt 2 lit. h oraz i, minister właściwy do spraw zdrowia niezwłocznie przekazuje Prezesowi Agencji, w celu przygotowania:44)
Wniosek, o którym mowa w art. 24 ust. 1 pkt 1, dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który nie ma odpowiednika refundowanego w danym wskazaniu, wraz z analizami, o których mowa w art. 25 pkt 14 lit. c, a także wniosek, o którym mowa w art. 24 ust. 1 pkt 2, dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który nie ma odpowiednika refundowanego w danym wskazaniu, jeżeli w uzasadnieniu wniosku są podane argumenty związane z efektem zdrowotnym, dodatkowym efektem zdrowotnym lub kosztami ich uzyskania wraz z analizami, o których mowa w art. 26 pkt 2 lit. h oraz i, minister właściwy do spraw zdrowia niezwłocznie przekazuje Prezesowi Agencji za pomocą SOLR, w celu przygotowania:45)
1) analizy weryfikacyjnej Agencji;
2) stanowiska Rady Przejrzystości;
3) rekomendacji Prezesa Agencji.
2.46) W przypadku stwierdzenia, że wniosek nie spełnia wymagań określonych w przepisach wydanych na podstawie art. 24 ust. 7, Prezes Agencji informuje o tym fakcie ministra właściwego do spraw zdrowia. Przepis art. 31 ust. 4 i 5 stosuje się.
2.47) W przypadku stwierdzenia, że wniosek nie spełnia wymagań określonych w przepisach wydanych na podstawie art. 24 ust. 7 pkt 2, Prezes Agencji wzywa wnioskodawcę do uzupełnienia analiz za pomocą SOLR, wyznaczając mu 21 dni na uzupełnienie dokumentacji. Bieg terminów, o których mowa w art. 31 ust. 4 i 5, ulega zawieszeniu.
3. Analiza weryfikacyjna Agencji podlega opłacie. Opłatę wnosi się na rachunek bankowy Agencji. Opłata wynosi nie więcej niż 150 000 zł.
4. Prezes Agencji niezwłocznie przekazuje analizę weryfikacyjną Agencji w sprawie oceny leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego Radzie Przejrzystości oraz wnioskodawcy, a następnie publikuje ją w Biuletynie Informacji Publicznej Agencji wraz z analizami wnioskodawcy, o których mowa w art. 25 pkt 14 lit. c oraz art. 26 pkt 2 lit. h oraz i. Do tych analiz można zgłaszać uwagi w terminie 7 dni od dnia opublikowania.
5. Analiza weryfikacyjna Agencji zawiera w szczególności:
1) ocenę analiz, o których mowa w art. 25 pkt 14 lit. c albo art. 26 pkt 2 lit. h oraz i;
2) przedstawienie rekomendacji refundacyjnych odnośnie wnioskowanego leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego z innych państw wraz z analizą ich uzasadnień i szczegółowych warunków objęcia refundacją;
3) warunki objęcia refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego w innych państwach z analizą szczegółowych warunków objęcia refundacją;
4) wyznaczenie wartości progowej ceny zbytu netto, przy której stosunek kosztów do uzyskiwanych efektów zdrowotnych nie jest większy od progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość, o którym mowa w art. 12 pkt 13 i art. 19 ust. 2 pkt 7, a w przypadku braku możliwości wyznaczenia tego kosztu – kosztu uzyskania dodatkowego roku życia.
6. Prezes Agencji, na podstawie stanowiska Rady Przejrzystości w sprawie oceny leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, przygotowuje rekomendację w zakresie:
1) objęcia refundacją danego leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego albo
2) niezasadności objęcia refundacją danego leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego.
7. Rekomendacja Prezesa Agencji zawiera w szczególności:
1) rozstrzygnięcie, czy lek, środek spożywczy specjalnego przeznaczenia żywieniowego oraz wyrób medyczny powinien być finansowany ze środków publicznych;
2) określenie szczegółowych warunków objęcia refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego oraz wyrobu medycznego w zakresie:
a) wskazań, w których lek, środek spożywczy specjalnego przeznaczenia żywieniowego oraz wyrób medyczny ma być objęty refundacją,
b) sugerowanego poziomu odpłatności, o którym mowa w art. 14,
c) sugestie co do włączenia do istniejącej lub utworzenia nowej grupy limitowej, o której mowa w art. 15,
d) uwagi i propozycje do opisu programu lekowego, jeżeli dotyczy,
e) propozycje instrumentów dzielenia ryzyka, o których mowa w art. 11 ust. 5;
3) uzasadnienie zawierające:
a) wskazanie dowodów naukowych, na podstawie których została wydana rekomendacja, w tym dotyczących:
– skuteczności klinicznej i praktycznej,
– bezpieczeństwa stosowania,
– stosunku kosztów do uzyskiwanych efektów zdrowotnych dotychczas refundowanych leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, w porównaniu z wnioskowanym,
– relacji korzyści zdrowotnych do ryzyka stosowania,
b) wskazanie istnienia alternatywnej technologii medycznej oraz jej efektywności klinicznej i bezpieczeństwa stosowania,
c) omówienie wpływu na wydatki podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych i świadczeniobiorców,
d) wskazanie i omówienie rekomendacji klinicznych oraz dotyczących finansowania ze środków publicznych danego leku, środka spożywczego specjalnego przeznaczenia żywieniowego w innych krajach,
e) wskazanie wartości progowej ceny zbytu netto, przy której stosunek kosztów do uzyskiwanych efektów zdrowotnych nie jest większy od progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość, o którym mowa w art. 12 pkt 13 i art. 19 ust. 2 pkt 7, a w przypadku braku możliwości wyznaczenia tego kosztu – kosztu uzyskania dodatkowego roku życia,
f) wskazanie czy zachodzą okoliczności, o których mowa w art. 13 ust. 3 oraz maksymalnego poziomu ceny ustalonego zgodnie z art. 13 ust. 4.
8.48) Prezes Agencji przekazuje ministrowi właściwemu do spraw zdrowia rekomendację, w terminie nie dłuższym niż 60 dni od dnia otrzymania dokumentów określonych w ust. 1.
8.49) Prezes Agencji przekazuje ministrowi właściwemu do spraw zdrowia rekomendację za pomocą SOLR, w terminie nie dłuższym niż 60 dni od dnia otrzymania dokumentów określonych w ust. 1.
9.48) Minister właściwy do spraw zdrowia przekazuje Komisji wniosek, o którym mowa w ust. 1, wraz z analizą weryfikacyjną Agencji, stanowiskiem Rady Przejrzystości, rekomendacją Prezesa Agencji oraz innymi dokumentami na podstawie których przygotowana została rekomendacja, celem przeprowadzenia negocjacji warunków objęcia refundacją.
9.49) Minister właściwy do spraw zdrowia przekazuje Komisji, za pomocą SOLR, wniosek, o którym mowa w ust. 1, wraz z analizą weryfikacyjną Agencji, stanowiskiem Rady Przejrzystości, rekomendacją Prezesa Agencji oraz innymi dokumentami, na podstawie których przygotowana została rekomendacja, celem przeprowadzenia negocjacji warunków objęcia refundacją.
10. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia:
1) sposób i procedury przygotowania analizy weryfikacyjnej Agencji uwzględniając wiedzę z zakresu oceny technologii medycznych;
2) wysokość opłat, o których mowa w ust. 3, mając na uwadze przedmiot analizy oraz koszty przygotowania analizy weryfikacyjnej Agencji.
Art. 36.50) Wniosek, o którym mowa w art. 24 ust. 1 pkt 1 dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który ma swój odpowiednik refundowany w danym wskazaniu, oraz wnioski, o których mowa w art. 24 ust. 1 pkt 2–5, minister właściwy do spraw zdrowia przekazuje Komisji, celem przeprowadzenia negocjacji.
Art. 36.51) Wniosek, o którym mowa w art. 24 ust. 1 pkt 1, dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który ma swój odpowiednik refundowany w danym wskazaniu, oraz wnioski, o których mowa w art. 24 ust. 1 pkt 2–5, minister właściwy do spraw zdrowia przekazuje Komisji za pomocą SOLR, celem przeprowadzenia negocjacji.
Art. 37. 1. Minister właściwy do spraw zdrowia ogłasza, w drodze obwieszczenia, wykazy refundowanych:
1) leków,
2) środków spożywczych specjalnego przeznaczenia żywieniowego,
3) wyrobów medycznych
– w stosunku do których wydano ostateczne decyzje administracyjne o objęciu refundacją albo ostateczne decyzje zmieniające, o których mowa w art. 16.
2. Obwieszczenie, o którym mowa w ust. 1, zawiera:
1) dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny;
2) kategorię dostępności refundacyjnej;
3) poziom odpłatności;
4) urzędową cenę zbytu;
5) cenę detaliczną;
6) wysokość limitu finansowania dla leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, o którym mowa w art. 6 ust. 1 pkt 1 albo informacyjną wysokość limitu finansowania dla leków, środków spożywczych specjalnego przeznaczenia żywieniowego, o których mowa w art. 6 ust. 1 pkt 2 i 3 dostosowaną do wielkości opakowania jednostkowego;
7) wysokość dopłaty świadczeniobiorcy;
8) grupę limitową;
9) termin wejścia w życie decyzji, o której mowa w art. 11 oraz okres jej obowiązywania.
2a.52) Leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne przysługujące świadczeniobiorcom, o których mowa w art. 43a ust. 1 ustawy o świadczeniach, minister właściwy do spraw zdrowia ogłasza w obwieszczeniu, o którym mowa w ust. 1.
3. W przypadku kategorii, o której mowa w art. 6 ust. 1 pkt 2, opis programu lekowego stanowi załącznik do obwieszczenia, o którym mowa w ust. 1.
4. Minister właściwy do spraw zdrowia ogłasza, w drodze obwieszczenia, wykazy:
1) leków,
2) środków spożywczych specjalnego przeznaczenia żywieniowego
– o których mowa w art. 6 ust. 1 pkt 4, w stosunku do których wydano ostateczną decyzję administracyjną o ustaleniu urzędowej ceny zbytu.
5. Obwieszczenie, o którym mowa w ust. 4, zawiera:
1) dane identyfikujące lek, środek spożywczy specjalnego przeznaczenia żywieniowego;
2) urzędową cenę zbytu.
6. Obwieszczenia, o których mowa w ust. 1 i 4, są ogłaszane raz na 2 miesiące w dzienniku urzędowym ministra właściwego do spraw zdrowia.
7. Minister właściwy do spraw zdrowia przynajmniej raz w roku przekazuje Komisji Europejskiej obwieszczenia, o których mowa w ust. 1 i 4, oraz wykaz leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, co do których wydano decyzję o zmianie decyzji o objęciu refundacją w zakresie podwyższenia urzędowej ceny zbytu, wraz z informacją o tych cenach.
8.53) Minister właściwy do spraw zdrowia przekazuje do systemu informacji w ochronie zdrowia, o którym mowa w ustawie z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia, dane objęte obwieszczeniem, o którym mowa w ust. 1, w terminie nie krótszym niż 10 dni przed dniem wejścia w życie obwieszczenia.
Art. 38. 1.54) Świadczeniobiorcom przysługuje, na zasadach określonych w ustawie, zaopatrzenie w wyroby medyczne, na zlecenie osoby uprawnionej, oraz ich naprawa. Kontynuacja zaopatrzenia w wyroby medyczne określone w przepisach wydanych na podstawie ust. 4 może odbywać się także na zlecenie pielęgniarki i położnej, o których mowa w art. 15a ust. 1 i 2 ustawy z dnia 15 lipca 2011 r. o zawodach pielęgniarki i położnej (Dz. U. z 2014 r. poz. 1435, z późn. zm.55)).
2. Udział środków publicznych w cenie wyrobu medycznego nie może być niższy niż kwota stanowiąca 50% jego limitu finansowania ze środków publicznych, określonego w przepisach wydanych na podstawie ust. 4.
3. Limit finansowania ze środków publicznych określony dla naprawy wyrobu medycznego może być wykorzystany przez świadczeniobiorcę do dokonania naprawy tego wyrobu w ustalonym dla niego okresie użytkowania. W przypadku dokonania naprawy i wykorzystaniu części lub całości tego limitu okres użytkowania wyrobu medycznego ulega wydłużeniu proporcjonalnie do wykorzystanej części limitu naprawy, z zaokrągleniem w dół do pełnego miesiąca.
4. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, wykaz wyrobów medycznych wydawanych na zlecenie:
1) osoby uprawnionej,
2) pielęgniarki i położnej
– z określeniem limitów ich finansowania ze środków publicznych i wysokości udziału własnego świadczeniobiorcy w tym limicie i kryteria ich przyznawania,
3) okresy użytkowania oraz limity cen ich napraw
– uwzględniając skuteczność i bezpieczeństwo ich stosowania, sposób ich wytwarzania, oraz możliwości płatnicze podmiotu zobowiązanego do finansowania świadczeń opieki zdrowotnej ze środków publicznych.
4a.56) Zlecenia na zaopatrzenie i zlecenia naprawy są wystawiane w postaci elektronicznej albo papierowej.
4b.56) Zlecenia w postaci papierowej wystawia się w przypadku:
1) braku dostępu do systemu teleinformatycznego, o którym mowa w art. 7 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia;
2) zlecenia dla osoby o nieustalonej tożsamości;
3) zlecenia, o którym mowa w art. 42b ust. 11 pkt 2 ustawy o świadczeniach.
5.57) Zlecenie na zaopatrzenie zawiera:
1) dane świadczeniobiorcy:
a) imię albo imiona i nazwisko albo oznaczenie „NN” w przypadku osób o nieustalonej tożsamości,
b) adres (nazwa miejscowości, ulica, numer domu, kod pocztowy, numer lokalu, jeżeli nadano):
– miejsca zamieszkania albo
– miejsca pełnienia służby wojskowej, jeżeli dotyczy, albo
– urzędu gminy lub gminnego ośrodka pomocy społecznej – w przypadku świadczeniobiorcy, wobec którego wydano decyzję, o której mowa w art. 54 ust. 1 ustawy o świadczeniach, albo świadczeniodawcy, który udzielił świadczenia opieki zdrowotnej, albo
– „NMZ” w przypadku osób o nieustalonym miejscu zamieszkania,
c) identyfikator usługobiorcy w rozumieniu art. 17c ust. 2 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia,
d) datę urodzenia pacjenta albo wiek pacjenta, w przypadku gdy numer PESEL nie został nadany albo jest nieustalony,
e) kod uprawnienia dodatkowego, jeżeli dotyczy,
f) rodzaj, numer, datę wydania, termin ważności dokumentu potwierdzającego uprawnienie dodatkowe, jeżeli dotyczy,
g) adres poczty elektronicznej – w przypadku zlecenia w postaci elektronicznej, jeżeli posiada,
h) numer telefonu – w przypadku zlecenia w postaci elektronicznej, jeżeli posiada,
i) adres korespondencyjny – w przypadku zlecenia w postaci elektronicznej, jeżeli posiada;
2) dane dotyczące podmiotu, w ramach którego wystawiono zlecenie:
a) w przypadku podmiotu wykonującego działalność leczniczą – nazwę albo firmę, łącznie z nazwą jednostki organizacyjnej, i nazwę komórki organizacyjnej, jeżeli dotyczy,
b) adres miejsca udzielania świadczenia opieki zdrowotnej, a w przypadku osób wykonujących działalność leczniczą wyłącznie w miejscu wezwania – adres miejsca przyjmowania wezwań i miejsca przechowywania dokumentacji medycznej,
c) numer telefonu dostępny w miejscu udzielania świadczenia opieki zdrowotnej, a w przypadku osób wykonujących działalność leczniczą wyłącznie w miejscu wezwania – numer telefonu kontaktowego do osoby wystawiającej zlecenie,
d) identyfikator miejsca udzielania świadczenia opieki zdrowotnej w rozumieniu art. 17c ust. 4 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia, jeżeli nadano;
3) dane dotyczące osoby wystawiającej zlecenie, które w przypadku zlecenia w postaci papierowej są nanoszone w sposób czytelny za pomocą nadruku, pieczątki lub naklejki przymocowanej do zlecenia w sposób uniemożliwiający jej usunięcie bez zniszczenia druku zlecenia:
a) imię albo imiona i nazwisko osoby wystawiającej zlecenie,
b) kwalifikacje zawodowe osoby wystawiającej zlecenie, w tym posiadany tytuł zawodowy i specjalizacja,
c) identyfikator pracownika medycznego, o którym mowa w art. 17c ust. 5 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia,
d) numer telefonu do bezpośredniego kontaktu z osobą wystawiającą zlecenie,
e) podpis, w przypadku zlecenia:
– w postaci elektronicznej składany za pomocą bezpiecznego podpisu elektronicznego weryfikowanego przy pomocy ważnego kwalifikowanego certyfikatu w rozumieniu ustawy z dnia 18 września 2001 r. o podpisie elektronicznym albo podpisu potwierdzonego profilem zaufanym ePUAP, albo
– w postaci papierowej – podpis własnoręczny;
4) dane dotyczące wyrobu medycznego:
a) określenie,
b) liczbę porządkową, zgodnie z przepisami wydanymi na podstawie ust. 4,
c) liczbę sztuk,
d) uzasadnienie zalecenia na zaopatrzenie danym wyrobem,
e) dodatkowe informacje istotne przy doborze:
– stronę zaopatrzenia, jeżeli dotyczy,
– dane dotyczące soczewek okularowych korekcyjnych, jeżeli dotyczy,
f) miesiąc, którego dotyczy zaopatrzenie comiesięczne, jeżeli dotyczy;
5) datę wystawienia zlecenia;
6) dodatkowe wskazania osoby wystawiającej zlecenie;
7) numer identyfikujący zlecenie nadawany przez system teleinformatyczny usługodawcy lub system, o którym mowa w art. 7 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia, a w przypadku zlecenia w postaci papierowej numer ewidencyjny zlecenia nadawany przez oddział wojewódzki Funduszu;
8) kod oddziału wojewódzkiego Funduszu, a w przypadku zleceń wydawanych na podstawie przepisów o koordynacji – kod kraju instytucji właściwej;
9) numer umowy o udzielanie świadczeń opieki zdrowotnej zawartej z oddziałem wojewódzkim Funduszu, jeżeli dotyczy;
10) sposób powiadomienia świadczeniobiorcy o potwierdzonym zleceniu na podstawie art. 38a ust. 7 – w przypadku zlecenia w postaci elektronicznej.
5a.58) Potwierdzenie uprawnienia do zaopatrzenia w wyroby medyczne zawiera:
1) numer identyfikujący potwierdzenie uprawnienia do zaopatrzenia w wyroby medyczne nadawany przez system teleinformatyczny Funduszu – w przypadku zlecenia w postaci elektronicznej;
2) numer identyfikujący zlecenie, o którym mowa w ust. 5 pkt 7;
3) dane dotyczące wyrobu medycznego:
a) określenie,
b) liczbę porządkową nadaną zgodnie z przepisami wydanymi na podstawie ust. 4,
c) liczbę sztuk,
d) stronę zaopatrzenia, jeżeli dotyczy;
4) nazwę i kod oddziału wojewódzkiego Funduszu;
5) limit finansowania ze środków publicznych ustalony zgodnie z przepisami wydanymi na podstawie ust. 4;
6) poziom refundacji określony w procentach;
7) uzasadnienie odmowy potwierdzenia prawa do refundacji, jeżeli dotyczy;
8) datę potwierdzenia albo odmowy potwierdzenia zlecenia;
9) podpis:
a) składany za pomocą bezpiecznego podpisu elektronicznego weryfikowanego przy pomocy ważnego kwalifikowanego certyfikatu w rozumieniu ustawy z dnia 18 września 2001 r. o podpisie elektronicznym albo za pomocą bezpiecznego podpisu elektronicznego weryfikowanego przy pomocy wydawanego przez Fundusz ważnego certyfikatu w rozumieniu ustawy z dnia 18 września 2001 r. o podpisie elektronicznym, albo podpisu potwierdzonego profilem zaufanym ePUAP – w przypadku zlecenia w postaci elektronicznej, albo
b) podpis własnoręczny i pieczątka oddziału wojewódzkiego Funduszu – w przypadku zlecenia w postaci papierowej.
5b.58) Realizacja zlecenia zawiera:
1) numer identyfikujący realizację zlecenia nadawany przez system teleinformatyczny usługodawcy lub system, o którym mowa w art. 7 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia – w przypadku zlecenia w postaci elektronicznej;
2) numer identyfikujący zlecenie, o którym mowa w ust. 5 pkt 7;
3) nazwę albo firmę realizatora, łącznie z nazwą i adresem miejsca realizacji zaopatrzenia;
4) identyfikator usługodawcy, o którym mowa w art. 17c ust. 3 pkt 3 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia;
5) numer umowy o udzielanie świadczeń opieki zdrowotnej podmiotu realizującego czynności z zakresu zaopatrzenia w wyroby medyczne zawartej z oddziałem wojewódzkim Funduszu;
6) dane dotyczące wyrobu medycznego:
a) liczbę porządkową, zgodnie z przepisami wydanymi na podstawie ust. 4,
b) liczbę sztuk,
c) wytwórcę, model, nazwę handlową,
d) cenę detaliczną,
e) kwotę refundacji,
f) wysokość dopłaty świadczeniobiorcy,
g) miesiąc, którego dotyczy zaopatrzenie przysługujące comiesięcznie, jeżeli dotyczy;
7) datę przyjęcia do realizacji;
8) datę realizacji (wydania);
9) imię i nazwisko osoby realizującej zlecenie;
10) imię i nazwisko osoby odbierającej;
11) numer PESEL, a w przypadku jego braku nazwę i numer dokumentu potwierdzającego tożsamość osoby odbierającej;
12) własnoręczny podpis osoby odbierającej.
5c.58) W przypadku zlecenia na zaopatrzenie w postaci elektronicznej dane określone w ust. 5b pkt 4–12 są nanoszone na wydruku potwierdzającym odbiór wyrobu medycznego.
5d.58) Karta potwierdzenia uprawnienia w postaci papierowej na zaopatrzenie w wyroby medyczne przysługujące comiesięcznie zawiera:
1) dane dotyczące świadczeniobiorcy:
a) imię albo imiona i nazwisko albo oznaczenie „NN” w przypadku osób o nieustalonej tożsamości,
b) adres (nazwa miejscowości, ulica, numer domu, kod pocztowy, numer lokalu, jeżeli nadano):
– miejsca zamieszkania albo
– miejsca pełnienia służby wojskowej, jeżeli dotyczy, albo
– urzędu gminy lub gminnego ośrodka pomocy społecznej – w przypadku świadczeniobiorcy, wobec którego wydano decyzję, o której mowa w art. 54 ust. 1 ustawy o świadczeniach, albo świadczeniodawcy, który udzielił świadczenia opieki zdrowotnej, albo
– „NMZ” w przypadku osób o nieustalonym miejscu zamieszkania,
c) identyfikator usługobiorcy w rozumieniu art. 17c ust. 2 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia,
d) datę urodzenia pacjenta albo wiek pacjenta, w przypadku gdy numer PESEL nie został nadany albo jest nieustalony;
2) dane dotyczące wyrobu medycznego:
a) określenie,
b) liczbę porządkową, zgodnie z przepisami wydanymi na podstawie ust. 4,
c) liczbę sztuk,
d) limit finansowania ze środków publicznych ustalony zgodnie z przepisami wydanymi na podstawie ust. 4,
e) poziom refundacji określony w procentach;
3) okres ważności karty: od (miesiąc, rok) – do (miesiąc, rok);
4) nazwę i kod oddziału wojewódzkiego Funduszu;
5) numer ewidencyjny karty potwierdzenia uprawnienia na zaopatrzenie w wyroby medyczne przysługujące comiesięcznie nadawany przez oddział wojewódzki Funduszu;
6) pieczątkę i podpis pracownika oddziału wojewódzkiego Funduszu.
5e.58) Potwierdzenie wystawionych zleceń w postaci papierowej na zaopatrzenie przysługujące comiesięcznie zawiera:
1) datę wystawienia zlecenia;
2) określenie wyrobu medycznego;
3) liczbę sztuk wyrobu medycznego;
4) miesiąc, którego dotyczy zaopatrzenie przysługujące comiesięcznie, jeżeli dotyczy;
5) datę realizacji (wydania) zlecenia;
6) pieczątkę i podpis własnoręczny osoby wystawiającej zlecenie;
7) pieczątkę i podpis własnoręczny osoby realizującej zlecenie.
6.59) Zlecenie naprawy zawiera:
1) dane dotyczące świadczeniobiorcy:
a) imię albo imiona i nazwisko albo oznaczenie „NN” w przypadku osób o nieustalonej tożsamości,
b) adres (nazwa miejscowości, ulica, numer domu, kod pocztowy, numer lokalu, jeżeli nadano):
– miejsca zamieszkania albo
– miejsca pełnienia służby wojskowej, jeżeli dotyczy, albo
– urzędu gminy lub gminnego ośrodka pomocy społecznej – w przypadku świadczeniobiorcy, wobec którego wydano decyzję, o której mowa w art. 54 ust. 1 ustawy o świadczeniach, albo świadczeniodawcy, który udzielił świadczenia opieki zdrowotnej, albo
– „NMZ” w przypadku osób o nieustalonym miejscu zamieszkania,
c) identyfikator usługobiorcy, o którym mowa w art. 17c ust. 2 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia,
d) datę urodzenia pacjenta albo wiek pacjenta, w przypadku gdy numeru PESEL nie został nadany albo jest nieustalony,
e) adres poczty elektronicznej, jeżeli posiada – w przypadku zlecenia w postaci elektronicznej,
f) numer telefonu – w przypadku zlecenia w postaci elektronicznej;
2) dane dotyczące wyrobu medycznego:
a) określenie (wytwórca, model, nazwa handlowa),
b) liczbę porządkową,
c) liczbę sztuk,
d) uzasadnienie naprawy;
3) numer identyfikujący zlecenie naprawy nadawany przez system teleinformatyczny Funduszu, a w przypadku zlecenia naprawy w postaci papierowej – numer ewidencyjny zlecenia nadawany przez oddział wojewódzki Funduszu;
4) potwierdzenie uprawnienia do naprawy wyrobu medycznego:
a) nazwę i kod oddziału wojewódzkiego Funduszu,
b) limit ceny naprawy,
c) datę potwierdzenia zlecenia;
5) odmowę potwierdzenia uprawnienia do naprawy wyrobu medycznego, jeżeli dotyczy:
a) przyczynę odmowy potwierdzenia,
b) datę odmowy potwierdzenia;
6) informację o przedłużeniu okresu użytkowania wyrobu medycznego;
7) podpis, w przypadku zlecenia:
a) w postaci elektronicznej – składany za pomocą bezpiecznego podpisu elektronicznego weryfikowanego przy pomocy ważnego kwalifikowanego certyfikatu w rozumieniu ustawy z dnia 18 września 2001 r. o podpisie elektronicznym albo za pomocą bezpiecznego podpisu elektronicznego weryfikowanego przy pomocy wydawanego przez Fundusz ważnego certyfikatu w rozumieniu ustawy z dnia 18 września 2001 r. o podpisie elektronicznym, albo podpisu potwierdzonego profilem zaufanym ePUAP, albo
b) w postaci papierowej – podpis własnoręczny i pieczątka oddziału wojewódzkiego Funduszu.
6a.60) Realizacja zlecenia naprawy zawiera:
1) numer identyfikujący realizację zlecenia naprawy nadawany przez system teleinformatyczny usługodawcy lub system, o którym mowa w art. 7 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia – w przypadku zlecenia w postaci elektronicznej;
2) numer identyfikujący zlecenie naprawy, o którym mowa w ust. 6 pkt 3;
3) nazwę albo firmę realizatora, łącznie z nazwą i adresem miejsca realizacji zlecenia naprawy;
4) identyfikator usługodawcy, o którym mowa w art. 17c ust. 3 ustawy z dnia 28 kwietnia 2011 r. o systemie informacji w ochronie zdrowia;
5) numer umowy o udzielanie świadczeń opieki zdrowotnej podmiotu realizującego czynności z zakresu zaopatrzenia w wyroby medyczne zawartej z oddziałem wojewódzkim Funduszu;
6) dane dotyczące wyrobu medycznego:
a) określenie,
b) wytwórcę, model, nazwę handlową,
c) opis przeprowadzonej naprawy wraz ze specyfikacją,
d) okres gwarancji,
e) cenę naprawy;
7) datę przyjęcia do realizacji;
8) datę realizacji (wydania);
9) imię i nazwisko osoby realizującej zlecenie naprawy;
10) imię i nazwisko osoby odbierającej;
11) numer PESEL, a w przypadku jego braku nazwę i numer dokumentu potwierdzającego tożsamość osoby odbierającej;
12) własnoręczny podpis osoby odbierającej.
6b.60) W przypadku zlecenia w postaci elektronicznej dane określone w ust. 6a pkt 1, 2 i 6–12, są nanoszone na wydruku potwierdzającym odbiór wyrobu medycznego.
7.61) Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia:
1) sposób i tryb realizacji zleceń w postaci elektronicznej i papierowej,
2) szczegółowy zakres danych zawartych w zleceniu w postaci elektronicznej i wzór zlecenia w postaci papierowej,
3) sposób przechowywania zleceń w postaci elektronicznej i papierowej,
4) sposób realizacji zleceń w postaci elektronicznej i w postaci papierowej, w tym autoryzacji zlecenia w postaci elektronicznej,
5) minimalne wymagania techniczne dla systemów teleinformatycznych, których spełnienie jest konieczne dla wystawiania i realizacji zleceń w postaci elektronicznej
– uwzględniając konieczność zapewnienia prawidłowego wystawiania zleceń oraz prawidłowej ich realizacji, a także mając na uwadze konieczność zapewnienia bezpieczeństwa i integralności danych.
8. Podmiot realizujący zaopatrzenie w zakresie wyrobów medycznych jest obowiązany, w ramach kontroli, o której mowa w art. 64 ustawy o świadczeniach, udostępnić podmiotowi zobowiązanemu do finansowania świadczeń ze środków publicznych, w terminie określonym przez ten podmiot, informacje o treści każdej umowy, a także inne dokumenty oraz uzgodnienia w jakiejkolwiek formie, pomiędzy podmiotem a dostawcą, których celem jest nabycie wyrobów medycznych.
9. Do wyrobów medycznych, o których mowa w ust. 1, nie stosuje się przepisów art. 3–11, art. 12 pkt 1–3, 7, 8 i 10–13, oraz art. 13–37.
10. W celu potwierdzenia zrealizowania zaopatrzenia w zakresie wyrobów medycznych, Fundusz może zwrócić się do świadczeniobiorcy o informację dotyczącą udzielonego mu świadczenia przez świadczeniodawcę.
Art. 38a.62) 1. W przypadku wystawienia zlecenia w postaci elektronicznej świadczeniobiorca otrzymuje informację o wystawionym zleceniu zawierającą następujące dane:
1) klucz dostępu;
2) kod dostępu;
3) identyfikator zlecenia;
4) rodzaj zlecenia;
5) datę wystawienia zlecenia;
6) miesiąc, na który wystawiono zlecenie, jeżeli dotyczy;
7) imię i nazwisko świadczeniobiorcy;
8) imię i nazwisko osoby wystawiającej zlecenie;
9) numer prawa wykonywania zawodu osoby wystawiającej zlecenie, w tym posiadany tytuł zawodowy i specjalizację;
10) numer telefonu do bezpośredniego kontaktu z osobą wystawiającą zlecenie;
11) liczbę porządkową wyrobu medycznego;
12) liczbę sztuk wyrobu medycznego;
13) określenie wyrobu medycznego.
2. Informację, o której mowa w ust. 1, świadczeniobiorca otrzymuje:
1) na wskazany w systemie informacji w ochronie zdrowia adres poczty elektronicznej;
2) na wskazany w systemie informacji w ochronie zdrowia numer telefonu, w formie wiadomości tekstowej zawierającej kod dostępu, o którym mowa w ust. 1 pkt 2, oraz informację o wymagalności numeru dokumentu potwierdzającego tożsamość przy realizacji zlecenia;
3) w przypadku braku wskazania w systemie informacji w ochronie zdrowia danych, o których mowa w pkt 1 i 2, oraz na każde żądanie świadczeniobiorcy, w formie wydruku, a w przypadku udzielania świadczenia zdrowotnego w miejscu wezwania i braku możliwości przekazania informacji w formie wydruku – w innej uzgodnionej formie zawierającej kod dostępu i nazwę wyrobu medycznego.
3. Informację przekazywaną w sposób, o którym mowa w ust. 2 pkt 3, przekazuje osoba wystawiająca zlecenie.
4. Informacja, o której mowa w ust. 1, nie może zawierać żadnych innych treści, w szczególności o charakterze reklamowym.
5. Informacja, o której mowa w ust. 1, nie zastępuje zlecenia.
6. W przypadku wystawienia zlecenia na zaopatrzenie w postaci elektronicznej osoba wystawiająca przekazuje to zlecenie w systemie teleinformatycznym do Funduszu w celu potwierdzenia uprawnień świadczeniobiorcy.
7. Fundusz, po weryfikacji uprawnień świadczeniobiorcy, przekazuje świadczeniobiorcy powiadomienie o potwierdzonym zleceniu zgodnie z wybranym przez świadczeniobiorcę sposobem:
1) na wskazany w systemie informacji w ochronie zdrowia adres poczty elektronicznej;
2) na wskazany w systemie informacji w ochronie zdrowia numer telefonu;
3) na wskazany w zleceniu numer telefonu;
4) na wskazany w zleceniu adres poczty elektronicznej;
5) na wskazany w zleceniu adres korespondencyjny;
6) osobiście we właściwym oddziale wojewódzkim Funduszu.
Art. 38b.62) Zlecenie naprawy wystawiane jest w postaci elektronicznej, za pośrednictwem systemu informacji w ochronie zdrowia, w przypadku wystawienia zlecenia na zaopatrzenie w postaci elektronicznej.
Art. 39. 1. Lek nieposiadający pozwolenia na dopuszczenie do obrotu na terytorium Rzeczypospolitej Polskiej i sprowadzany z zagranicy na warunkach i w trybie określonym w art. 4 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne oraz środek spożywczy specjalnego przeznaczenia żywieniowego, o którym mowa w art. 29a ustawy z dnia 25 sierpnia 2006 r. o bezpieczeństwie żywności i żywienia mogą być wydawane po wniesieniu przez świadczeniobiorcę opłaty ryczałtowej, o której mowa w art. 6 ust. 2 pkt 2, za opakowanie jednostkowe, pod warunkiem wydania zgody na refundację takich produktów przez ministra właściwego do spraw zdrowia.
2. Minister właściwy do spraw zdrowia rozpatruje wniosek o refundację produktów, o których mowa w ust. 1, w terminie nie dłuższym niż 30 dni od dnia wystąpienia o ich refundację przez świadczeniobiorcę.
3. W celu zbadania zasadności wydawania zgód na refundację produktów, o których mowa w ust. 1, minister właściwy do spraw zdrowia może wystąpić do Prezesa Agencji w trybie określonym w art. 31e ust. 1 ustawy o świadczeniach.
4. Minister właściwy do spraw zdrowia, uwzględniając rekomendację, o której mowa w art. 31h ust. 3 ustawy o świadczeniach, może umieścić produkty, o których mowa w ust. 1, w wykazie, o którym mowa w ust. 5.
5. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, wykaz leków i środków spożywczych specjalnego przeznaczenia żywieniowego, które nie mogą być refundowane w trybie, o którym mowa w ust. 1, mając na uwadze rekomendację Prezesa Agencji oraz zapewnienie bezpieczeństwa zdrowotnego pacjentów.
Art. 40. 1. Jeżeli jest to niezbędne dla ratowania życia i zdrowia świadczeniobiorców, w przypadku braku innych możliwych do zastosowania w danym stanie klinicznym procedur medycznych finansowanych ze środków publicznych, minister właściwy do spraw zdrowia, po zasięgnięciu opinii Rady Przejrzystości oraz konsultanta krajowego w odpowiedniej dziedzinie medycyny, może wydać z urzędu, przy uwzględnieniu:
1) kryteriów, o których mowa w art. 12 pkt 4–6, 9, 10, 12 i 13,
2) stosunku kosztów do uzyskiwanych efektów zdrowotnych
– decyzję administracyjną o objęciu refundacją leku przy danych klinicznych, w zakresie wskazań do stosowania lub dawkowania, lub sposobu podawania odmiennych niż określone w Charakterystyce Produktu Leczniczego w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne.
2. Opinię, o której mowa w ust. 1, Rada Przejrzystości wydaje w terminie 14 dni biorąc pod uwagę w szczególności istotność stanu klinicznego, w którym ma być stosowany lek.
Rozdział 6
Apteki i osoby uprawnione
Art. 41. 1. W celu realizacji świadczeń, o których mowa w art. 15 ust. 2 pkt 14, 17 i 18 ustawy o świadczeniach, podmiot prowadzący aptekę zawiera umowę z Funduszem na wydawanie refundowanego leku, środka spożywczego specjalnego przeznaczenia żywieniowego oraz wyrobu medycznego na receptę, zwaną dalej „umową na realizację recept”.
2. Umowa na realizację recept określa w szczególności:
1) imię i nazwisko osoby będącej kierownikiem apteki;
2) wskazanie adresu prowadzenia apteki;
3) zobowiązanie do stosowania limitów, cen oraz odpłatności i dopłat świadczeniobiorcy w wysokości określonej w obwieszczeniu, o którym mowa w art. 37;
4) kary umowne;
5) warunki jej wypowiedzenia albo rozwiązania.
3. Umowa na realizację recept jest zawierana odrębnie dla każdej apteki na czas nieokreślony. Umowa podpisywana jest również przez kierownika apteki.
4. W celu zawarcia umowy na realizację recept podmiot prowadzący aptekę przedstawia następujące dokumenty:
1) kopię zezwolenia na prowadzenie apteki;
2) kopie dokumentów uprawniających kierownika apteki do pełnienia tej funkcji;
3) aktualną ewidencję osób zatrudnionych w aptece wraz z numerami dokumentów uprawniających do wykonywania zawodu;
4) numer rachunku bankowego podmiotu prowadzącego aptekę.
4a.63) W celu obsługi umów na realizację recept, Fundusz jest uprawniony do przetwarzania danych osobowych osób, o których mowa w ust. 4 pkt 3.
5. Fundusz nie może odmówić zawarcia umowy na realizację recept, z zastrzeżeniem ust. 7. Do zawierania umowy nie stosuje się przepisów o zamówieniach publicznych.
6. Fundusz rozwiązuje umowę na realizację recept ze skutkiem natychmiastowym w przypadku:
1) uniemożliwiania czynności kontrolnych;
2) niewykonania w terminie zaleceń pokontrolnych.
7. Fundusz nie zawiera kolejnej umowy przez okres:
1) jednego roku w przypadku pierwszego rozwiązania umowy, o którym mowa w ust. 6;
2) trzech lat w przypadku drugiego rozwiązania umowy, o którym mowa w ust. 6.
8. Minister właściwy do spraw zdrowia określi, w drodze rozporządzenia, ogólne warunki umów na realizację recept oraz ramowy wzór umowy na realizację recept, kierując się koniecznością zapewnienia właściwej ich realizacji.
Art. 42. 1. Podmiotowi prowadzącemu aptekę, który zawarł umowę na realizację recept, przysługuje zażalenie na czynności dyrektora wojewódzkiego oddziału Funduszu dotyczące realizacji umowy.
2. Zażalenie, o którym mowa w ust. 1, składa się wraz z uzasadnieniem za pośrednictwem właściwego miejscowo oddziału wojewódzkiego Funduszu w terminie 14 dni od dnia dokonania czynności przez dyrektora oddziału wojewódzkiego Funduszu.
3. Zażalenie, o którym mowa w ust. 1, rozpatruje, w terminie 14 dni od dnia jego otrzymania, Prezes Funduszu.
4. Prezes Funduszu, uwzględniając zażalenie w części lub w całości, nakłada na dyrektora oddziału wojewódzkiego Funduszu obowiązek usunięcia stwierdzonych nieprawidłowości, w szczególności poprzez uchylenie czynności, której dotyczy zażalenie, i zawiadamia podmiot prowadzący aptekę w terminie 7 dni o uwzględnieniu zażalenia.
5. W przypadku nieuwzględnienia zażalenia przez Prezesa Funduszu w części lub w całości, podmiotowi prowadzącemu aptekę przysługuje wniosek o ponowne rozpatrzenie sprawy.
6. Wniosek, o którym mowa w ust. 5, podmiot prowadzący aptekę może złożyć do Prezesa Funduszu w terminie 14 dni od dnia otrzymania stanowiska Prezesa na złożone przez podmiot prowadzący aptekę zażalenie.
7. Wniosek o ponowne rozpatrzenie sprawy rozpatrywany jest przez Prezesa Funduszu w terminie 14 dni od dnia jego otrzymania. Stanowisko Prezesa jest ostateczne.
Art. 43. 1. Apteka w celu realizacji świadczeń objętych umową na realizację recept ma obowiązek:
1) zapewnić świadczeniobiorcy dostępność leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, objętych wykazami o których mowa w art. 37;
2) gromadzić i przekazywać Funduszowi, rzetelne i zgodne ze stanem faktycznym na dzień przekazania, informacje zawarte w treści poszczególnych zrealizowanych recept na refundowane leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne;
3) udostępniać do kontroli prowadzonej przez podmiot zobowiązany do finansowania świadczeń ze środków publicznych dokumentację, którą apteka jest obowiązana prowadzić w związku z realizacją umowy, i udzielać wyjaśnień w zakresie związanym z realizacją recept podlegających refundacji;
4) udostępnić do kontroli podmiotowi zobowiązanemu do finansowania świadczeń opieki zdrowotnej ze środków publicznych, w terminie określonym przez ten podmiot, informacje o treści każdej umowy, w tym także uzgodnienia dokonanego w jakiejkolwiek formie, pomiędzy apteką a hurtownią farmaceutyczną, których celem jest nabycie leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych;
5) zamieścić, w widocznym i łatwo dostępnym miejscu, informację o zawarciu umowy na realizację recept oraz informację, o której mowa w art. 44 ust. 1;
6)64) zwrotu refundacji ceny leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego łącznie z odsetkami ustawowymi za opóźnienie liczonymi od dnia, w którym wypłacono refundację do dnia jej zwrotu, w terminie 14 dni od dnia otrzymania wezwania do zapłaty, jeżeli w wyniku weryfikacji lub kontroli informacji, o których mowa w pkt 2, lub kontroli, o której mowa w pkt 3 i 4, zostanie stwierdzone, że realizacja recepty nastąpiła z naruszeniem przepisów ustawy, przepisów o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych oraz przepisów o zawodach lekarza i lekarza dentysty;
7) przechowywać recepty na refundowane leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne wraz z otaksowaniem przez okres 5 lat, liczonych od zakończenia roku kalendarzowego, w którym nastąpiła refundacja.
2. W razie stwierdzenia w trakcie czynności kontrolnych nieprawidłowości innych niż naruszenie obowiązków, o których mowa w ust. 1, podmiot zobowiązany do finansowania świadczeń ze środków publicznych niezwłocznie powiadamia o tym fakcie wojewódzkiego inspektora farmaceutycznego.
3. W przypadku stwierdzenia w wyniku kontroli, o której mowa w ust. 1 pkt 4, naruszenia przepisów art. 49, podmiot zobowiązany do finansowania świadczeń ze środków publicznych niezwłocznie powiadamia o tym fakcie ministra właściwego do spraw zdrowia oraz wojewódzkiego inspektora farmaceutycznego.
Art. 44. 1. Osoba wydająca leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne objęte refundacją ma obowiązek poinformować świadczeniobiorcę o możliwości nabycia leku objętego refundacją, innego niż lek przepisany na recepcie, o tej samej nazwie międzynarodowej, dawce, postaci farmaceutycznej, która nie powoduje powstania różnic terapeutycznych, i o tym samym wskazaniu terapeutycznym, którego cena detaliczna nie przekracza limitu finansowania ze środków publicznych oraz ceny detalicznej leku przepisanego na recepcie. Apteka ma obowiązek zapewnić dostępność tego leku.
2.65) Osoba wydająca leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne objęte refundacją ma obowiązek, na żądanie świadczeniobiorcy, wydać lek, o którym mowa w ust. 1, którego cena detaliczna jest niższa niż cena leku przepisanego na recepcie.
2a.66) Osoba wydająca leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne objęte refundacją ma obowiązek, na żądanie świadczeniobiorcy, po uprzednim spełnieniu obowiązku, o którym mowa w ust. 1, wydać lek inny niż lek przepisany na recepcie, o tej samej nazwie międzynarodowej, dawce, postaci farmaceutycznej, która nie powoduje powstania różnic terapeutycznych, i o tym samym wskazaniu terapeutycznym, którego cena detaliczna jest równa lub wyższa od ceny leku przepisanego na recepcie.
2b.66) Osoba wydająca leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne objęte refundacją może na żądanie świadczeniobiorcy, po uprzednim spełnieniu obowiązku, o którym mowa w ust. 1, wydać lek nieobjęty refundacją, inny niż lek przepisany na recepcie, o tej samej nazwie międzynarodowej, dawce oraz o postaci farmaceutycznej, która nie powoduje powstania różnic terapeutycznych, za 100% odpłatnością.
2c.66) Osoba wydająca leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne objęte refundacją może wydać lek w opakowaniu zawierającym różną – o maksymalnie 10%, liczbę dawek niż określona na recepcie.
2d.66) Przepisy ust. 2–2c nie dotyczą sytuacji, w której osoba uprawniona umieściła odpowiedni wpis w recepcie – w przypadku recepty w postaci elektronicznej, lub adnotację na druku recepty – w przypadku recepty w postaci papierowej, wskazując na niemożność dokonania zamiany przepisanego leku.
3.67) Przepisy ust. 1–2d stosuje się odpowiednio do środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych.
Art. 44a.68) 1. W przypadku recepty wystawionej dla świadczeniobiorcy, który ukończył 75. rok życia, przez lekarza podstawowej opieki zdrowotnej, pielęgniarkę podstawowej opieki zdrowotnej albo lekarza posiadającego prawo wykonywania zawodu, który zaprzestał wykonywania zawodu i wystawił receptę dla siebie albo dla małżonka, wstępnych lub zstępnych w linii prostej oraz rodzeństwa, osoba wydająca leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne, oprócz obowiązków wynikających z art. 44, ma obowiązek poinformować o możliwości nabycia leku innego niż lek przepisany na recepcie, o tej samej nazwie międzynarodowej, dawce, postaci farmaceutycznej, która nie powoduje powstania różnic terapeutycznych, i o tym samym wskazaniu terapeutycznym, objętego wykazem, o którym mowa w art. 37 ust. 1, w części dotyczącej bezpłatnego zaopatrzenia świadczeniobiorców po ukończeniu 75. roku życia oraz ma obowiązek na żądanie wydać ten lek.
2. W przypadku zbiegu uprawnień, o których mowa w art. 43–45 ustawy o świadczeniach oraz w art. 7a ust. 1 pkt 2 ustawy z dnia 19 czerwca 1997 r. o zakazie stosowania wyrobów zawierających azbest (Dz. U. z 2004 r. poz. 20, 959, 1252 i 2135, z 2005 r. poz. 72 oraz z 2009 r. poz. 106), osoba wydająca leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne ma obowiązek wydać lek lub środek spożywczy specjalnego przeznaczenia żywieniowego z najniższą wysokością dopłaty.
3. Przepis ust. 1 nie dotyczy sytuacji, w której osoba uprawniona umieściła odpowiedni wpis w recepcie – w przypadku recepty w postaci elektronicznej, lub adnotację na druku recepty – w przypadku recepty w postaci papierowej, wskazując na niemożność dokonania zamiany przepisanego leku.
4. Przepisy art. 44 ust. 1–2d stosuje się odpowiednio.
Art. 45. 1. Apteki gromadzą informacje zawierające dane o obrocie lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi objętymi refundacją, wynikające ze zrealizowanych recept wystawionych przez osobę uprawnioną.
2. Dane są gromadzone i przechowywane w formie elektronicznej oraz przekazywane w formie komunikatów elektronicznych, oddziałowi wojewódzkiemu Funduszu, właściwemu ze względu na siedzibę apteki.
3. Przekazywanie komunikatów elektronicznych obejmuje następujące fazy:
1) przygotowanie komunikatu przez aptekę, sprawdzenie jego kompletności i poprawności oraz jego terminowe przekazanie;
2) weryfikację komunikatu przez Fundusz oraz przygotowanie i przekazanie komunikatu zwrotnego zawierającego wynik wstępnej analizy poprawności danych oraz projektu zestawienia zbiorczego;
3) poprawianie lub usuwanie przez aptekę, w drodze korekty, błędów albo innych nieprawidłowości wykazanych w komunikacie zwrotnym i ponowne przekazanie komunikatu elektronicznego.
4. Zatwierdzenie przez aptekę projektu zestawienia zbiorczego powoduje udostępnienie aptece komunikatu elektronicznego zawierającego uzgodnione zestawienie zbiorcze i zamyka dany okres rozliczeniowy oraz uniemożliwia aptece składanie do niego korekt w trybie określonym w ust. 3.
5. Apteki przekazują do właściwego oddziału wojewódzkiego Funduszu uzgodnione zestawienie zbiorcze w formie pisemnej, stanowiące podstawę refundacji.
6. Apteki przekazują dane w następujących terminach:
1) za okres od 1 do 15 dnia danego miesiąca – do pięciu dni roboczych od dnia zakończenia okresu rozliczeniowego;
2) za okres od 16 dnia do końca miesiąca – do pięciu dni roboczych od dnia zakończenia okresu rozliczeniowego.
7. Oddział wojewódzki Funduszu przeprowadza czynności, o których mowa w ust. 3 pkt 2, w terminie 5 dni roboczych od dnia otrzymania danych, o których mowa w ust. 3 pkt 1.
8. Czynność, o której mowa w ust. 3 pkt 3, apteka wykonuje jednorazowo w terminie 5 dni roboczych od otrzymania danych, o których mowa w ust. 3 pkt 2, po czym oddział wojewódzki Funduszu ponownie przeprowadza czynności, o których mowa w ust. 3 pkt 2, w terminie 5 dni roboczych od dnia otrzymania poprawionych danych.
9. Nieprzekazanie przez aptekę danych sporządzonych zgodnie z ust. 8, traktowane jest jako zatwierdzenie przez aptekę projektu zestawienia zbiorczego, powoduje udostępnienie aptece komunikatu elektronicznego zawierającego uzgodnione zestawienie zbiorcze, zamyka dany okres rozliczeniowy oraz uniemożliwia aptece składanie do niego korekt w trybie określonym w ust. 3.
10. Za datę złożenia uzgodnionego zestawienia zbiorczego w formie pisemnej przyjmuje się datę wpływu zestawienia do właściwego oddziału wojewódzkiego Funduszu.
11. Złożenie przez aptekę korekty do zamkniętego okresu rozliczeniowego jest możliwe jedynie po pozytywnym rozpatrzeniu umotywowanego wniosku apteki przez dyrektora oddziału wojewódzkiego Funduszu.
12. Korekty do zamkniętego okresu rozliczeniowego apteka może składać w terminie 3 miesięcy od daty jego zamknięcia, nie później jednak niż do 15 marca roku następnego, z zastrzeżeniem ust. 9.
13. Fundusz może prowadzić analizy uzgodnionych zestawień zbiorczych po zamknięciu okresu rozliczeniowego, co może skutkować wezwaniem do złożenia przez aptekę korekty.
14. Minister właściwy do spraw zdrowia, po zasięgnięciu opinii Prezesa Funduszu, określi, w drodze rozporządzenia, zakres niezbędnych informacji gromadzonych przez apteki, sposób ich rejestrowania oraz zakres informacji i sposób ich przekazywania Funduszowi, w tym także rodzaje wykorzystywanych nośników informacji oraz wzory komunikatów i dokumentów, biorąc pod uwagę zakres zadań wykonywanych przez te podmioty.
Art. 46. 1. Podmiot prowadzący aptekę, który zawarł umowę na realizację recept, po przedstawieniu właściwemu oddziałowi wojewódzkiemu Funduszu zestawienia, o którym mowa w art. 45 ust. 5, nie częściej niż co 14 dni, otrzymuje refundację ustalonego limitu finansowania leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego.
2.69) Refundacja nie może przekraczać ustalonego limitu finansowania, z zastrzeżeniem art. 43a–46 ustawy o świadczeniach.
3. Właściwy oddział wojewódzki Funduszu dokonuje refundacji nie później niż 15 dni od dnia otrzymania zestawienia, o którym mowa w art. 45 ust. 5.
4. W przypadku gdy złożone do Funduszu zestawienie jest niezgodne z komunikatem, o którym mowa w art. 45 ust. 4, Fundusz zwraca je aptece w terminie 7 dni od daty złożenia.
5. Zestawienie, o którym mowa w ust. 4, nie stanowi podstawy do dokonania przez Fundusz refundacji.
6.70) W przypadku przekroczenia przez Fundusz terminu, o którym mowa w ust. 3, aptece przysługują odsetki ustawowe za opóźnienie.
7.70) W przypadku stwierdzenia w wyniku analizy, o której mowa w art. 45 ust. 13, nienależnego obciążenia refundacją, Funduszowi przysługuje zwrot nienależnie wypłaconej refundacji wraz z odsetkami ustawowymi za opóźnienie liczonymi od dnia jej przekazania do dnia jej zwrotu.
8. Fundusz przekazuje dane, o których mowa w art. 45, wynikające ze zrealizowanych recept podlegających refundacji z budżetu państwa właściwym ministrom w terminie 30 dni od dnia ich otrzymania.
Art. 47. 1. Apteka jest obowiązana udostępnić, na żądanie Funduszu, do kontroli recepty wraz z ich otaksowaniem i przekazać niezbędne dane, o których mowa w art. 45 ust. 1, a także dokumentację, którą apteka jest zobowiązana prowadzić i posiadać na podstawie odrębnych przepisów, i informacje, o których mowa w art. 43 ust. 1 pkt 2–4.
2. Osobą uprawnioną do reprezentowania apteki w trakcie kontroli jest kierownik apteki lub upoważniony przez niego farmaceuta wyznaczony do zastępowania kierownika apteki, w trybie przepisów ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, a w przypadku gdy kierownikiem punktu aptecznego jest technik farmaceutyczny wyznaczony do jego zastępowania – technik farmaceutyczny.
3. Wydanie recept, o których mowa w ust. 1, wraz z otaksowaniem może nastąpić nie wcześniej niż po zamknięciu okresu rozliczeniowego, o którym mowa w art. 45 ust. 9, i na czas niezbędny do przeprowadzenia kontroli.
4. Wydanie recept i dokumentacji oraz udostępnienie informacji, o których mowa w ust. 1, odbywa się na podstawie pisemnego potwierdzenia przejęcia, określającego liczbę, rodzaj oraz numery recept nadane przez Fundusz zgodnie z przepisami wydanymi na podstawie art. 45 ust. 5 ustawy z dnia 5 grudnia 1996 r. o zawodach lekarza i lekarza dentysty (Dz. U. z 2015 r. poz. 464, z późn. zm.71)), podpisanego przez kontrolera i kierownika apteki lub osobę upoważnioną do zastępowania, o której mowa w ust. 2.
5. Kontrolę przeprowadza się w aptece lub w siedzibie oddziału Funduszu, jeżeli kontrola przeprowadzana jest na podstawie danych elektronicznych przesyłanych przez apteki w trybie art. 43 ust. 1 pkt 2 oraz art. 45 ust. 1 i 2.
6. Kontrolę, o której mowa w ust. 1, przeprowadza się na podstawie imiennego upoważnienia, udzielonego przez Fundusz, zawierającego:
1) wskazanie podstawy prawnej;
2) datę i miejsce wystawienia;
3) imię i nazwisko upoważnionej osoby, zwanej dalej „kontrolerem”;
4) oznaczenie kontrolowanej apteki, w tym numer zezwolenia na prowadzenie apteki;
5) wskazanie daty rozpoczęcia kontroli i przewidywanego terminu zakończenia kontroli;
6) zakres kontroli;
7) podpis oraz imienną pieczątkę osoby upoważnionej do reprezentowania Funduszu wraz z określeniem jej stanowiska służbowego;
8) pouczenie o prawach i obowiązkach kontrolowanej apteki.
7. W ramach udzielonego upoważnienia kontroler ma prawo do:
1) wstępu do pomieszczeń apteki;
2) dostępu do recept, danych, dokumentacji i informacji, o których mowa w ust. 1, oraz wglądu w nie;
2a)72) dostępu do recept, danych, dokumentacji i informacji w postaci elektronicznej oraz wglądu w nie za pośrednictwem systemu informacji w ochronie zdrowia;
3) żądania od kierownika apteki i osób zatrudnionych w aptece ustnych i pisemnych wyjaśnień dotyczących przedmiotu kontroli.
8. Kontroler może żądać sporządzenia niezbędnych do kontroli odpisów i kopii dokumentów, jak również zestawień i obliczeń sporządzonych na podstawie dokumentów.
9. Zgodność odpisów i kopii z oryginałami dokumentów oraz prawidłowość zestawień i obliczeń potwierdza kierownik apteki lub osoba upoważniona do zastępowania, o której mowa w ust. 2.
10. Po przeprowadzeniu kontroli kontroler sporządza protokół kontroli zawierający zbiór ustaleń dotyczących realizacji umowy przez aptekę. Kierownik apteki lub farmaceuta, o którym mowa w ust. 2, otrzymuje jeden egzemplarz protokołu kontroli.
11. Protokół kontroli podpisują:
1) kierownik apteki lub osoba upoważniona do zastępowania, o której mowa w ust. 2;
2) kontroler przeprowadzający kontrolę.
12. W przypadku odmowy podpisania protokołu kontroli odmawiający składa pisemne wyjaśnienie co do przyczyn odmowy, w terminie 7 dni od dnia jego doręczenia.
13. O odmowie podpisania protokołu kontroli oraz o przyczynie tej odmowy kontroler dokonuje wzmianki w protokole kontroli.
14. Kierownik apteki lub osoba upoważniona do zastępowania, o której mowa w ust. 2, jeżeli nie zgadza się z ustaleniami protokołu kontroli, może, w terminie 7 dni od dnia jego doręczenia, złożyć pisemnie zastrzeżenia, wskazując jednocześnie stosowne wnioski dowodowe. Kontroler jest obowiązany rozpatrzyć zgłoszone zastrzeżenia w terminie 7 dni od dnia ich otrzymania. W przypadku uwzględnienia zastrzeżeń kontroler uzupełnia protokół kontroli i przedstawia go ponownie do podpisu.
15. Na podstawie ustaleń zawartych w protokole kontroli oddział wojewódzki Funduszu wydaje zalecenia pokontrolne, zobowiązujące kierownika apteki oraz podmiot prowadzący aptekę do usunięcia, w określonym terminie, stwierdzonych uchybień oraz do złożenia, w terminie 14 dni od dnia doręczenia zaleceń pokontrolnych, informacji o podjętych działaniach. Od zaleceń pokontrolnych przysługuje kierownikowi apteki lub podmiotowi prowadzącemu aptekę zażalenie do dyrektora oddziału wojewódzkiego Funduszu. Zażalenie wnosi się w terminie 7 dni od dnia otrzymania zaleceń pokontrolnych. Dyrektor oddziału wojewódzkiego Funduszu rozpatruje zażalenie w terminie 14 dni od dnia jego otrzymania i w przypadku jego uwzględnienia zmienia zalecenia pokontrolne.
16. W przypadku nieuwzględnienia zażalenia, o którym mowa w ust. 15, w części lub w całości, kierownikowi apteki lub podmiotowi prowadzącemu aptekę przysługuje odwołanie do Prezesa Funduszu. Przepisy art. 42 ust. 6 i 7 stosuje się odpowiednio.
17. Wniesienie zażalenia lub odwołania wstrzymuje wykonanie zaleceń pokontrolnych.
Art. 48. 1.73) Realizacja świadczeń, o których mowa w art. 15 ust. 2 pkt 14, 17 i 18 ustawy o świadczeniach, przysługuje świadczeniobiorcy na podstawie recepty wystawionej przez osobę uprawnioną, o której mowa w art. 2 pkt 14. Realizacja świadczeń, o których mowa w art. 15 ust. 2 pkt 9 ustawy o świadczeniach, przysługuje świadczeniobiorcy na podstawie zlecenia wystawionego przez osobę uprawnioną, o której mowa w art. 2 pkt 14.
2. (uchylony)74)
2a.75) Recepty w postaci papierowej na refundowane leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne opatrzone są unikalnymi numerami identyfikującymi recepty, nadawanymi przez dyrektora oddziału wojewódzkiego Funduszu.
3. (uchylony)76)
4. (uchylony)76)
5.77) W przypadku prawomocnego skazania za przestępstwo określone w art. 54 ust. 2, 3 lub 5 ustawy lub art. 228–230, art. 286 lub art. 296a ustawy z dnia 6 czerwca 1997 r. – Kodeks karny, osoba uprawniona traci prawo wystawiania recept na leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne refundowane na okres:
1) jednego roku – w przypadku pierwszego skazania;
2) trzech lat – w przypadku kolejnego skazania.
6. (uchylony)78)
7.79) Osoba uprawniona, z wyłączeniem lekarza ubezpieczenia zdrowotnego, felczera ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, pielęgniarki ubezpieczenia zdrowotnego i położnej ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, o których mowa w art. 15a ust. 1 i 2 ustawy z dnia 15 lipca 2011 r. o zawodach pielęgniarki i położnej, oraz podmiot wykonujący działalność leczniczą, w ramach którego wystawiono receptę na co najmniej jeden refundowany lek, środek spożywczy specjalnego przeznaczenia żywieniowego oraz wyrób medyczny lub zlecenie na wyrób medyczny, są obowiązani poddać się kontroli przeprowadzanej lub zlecanej przez Fundusz w zakresie dokumentacji medycznej dotyczącej zasadności i prawidłowości wystawiania recept na refundowane leki, środki spożywcze specjalnego przeznaczenia żywieniowego oraz wyroby medyczne i zleceń na wyroby medyczne. Do kontroli stosuje się odpowiednio przepisy art. 64 ust. 1–10 ustawy o świadczeniach.
7a.80) Osoba uprawniona z wyłączeniem lekarza ubezpieczenia zdrowotnego, felczera ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, pielęgniarki ubezpieczenia zdrowotnego i położnej ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, o których mowa w art. 15a ust. 1 i 2 ustawy z dnia 15 lipca 2011 r. o zawodach pielęgniarki i położnej, jest obowiązana do zwrotu Funduszowi kwoty stanowiącej równowartość kwoty refundacji wraz z odsetkami ustawowymi liczonymi od dnia dokonania refundacji, o której mowa w art. 46 ust. 1, w przypadku:
1) wypisania recepty w okresie pozbawienia prawa wystawiania recept, o którym mowa w ust. 5;
2) wypisania recepty nieuzasadnionej udokumentowanymi względami medycznymi;
3) wypisania recepty niezgodnej z uprawnieniami świadczeniobiorcy;
4) wypisania recepty niezgodnej ze wskazaniami zawartymi w obwieszczeniach, o których mowa w art. 37 ust. 1 lub 4.
7b.80) Osoba uprawniona, z wyłączeniem lekarza ubezpieczenia zdrowotnego, felczera ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, pielęgniarki ubezpieczenia zdrowotnego i położnej ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, o których mowa w art. 15a ust. 1 i 2 ustawy z dnia 15 lipca 2011 r. o zawodach pielęgniarki i położnej, jest obowiązana do zwrotu Funduszowi kwoty stanowiącej równowartość limitu finansowania wyrobu medycznego wraz z odsetkami ustawowymi liczonymi od dnia dokonania finansowania, o którym mowa w przepisach wydanych na podstawie art. 38 ust. 4, w przypadku:
1) wypisania zlecenia w okresie pozbawienia prawa wystawiania recept, o którym mowa w ust. 5;
2) wypisania zlecenia nieuzasadnionego udokumentowanymi względami medycznymi;
3) wypisania zlecenia niezgodnego z kryteriami przyznawania zawartymi w przepisach, o których mowa w art. 38 ust. 4.
8. (uchylony)
9. (uchylony)
10. (uchylony)81)
11. (uchylony)81)
Art. 49. 1. Zakazuje się przedsiębiorcy zajmującemu się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi podlegającymi refundacji:
1) uzależniania zawarcia umowy dotyczącej tych leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych lub uzależniania treści tej umowy od przyjęcia lub spełnienia przez:
a) innego przedsiębiorcę zajmującego się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi podlegającymi refundacji,
b) podmiot prowadzący aptekę,
c) kierownika apteki,
d) osobę wydającą leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne objęte refundacją
– świadczenia niezwiązanego z przedmiotem tej umowy, w tym korzyści majątkowej lub osobistej, o której mowa w ust. 3;
2) stosowania wobec podmiotów wymienionych w pkt 1 niejednolitych warunków umów.
2. Umowy sprzeczne z ust. 1 są w tym zakresie nieważne.
3. Zakazuje się:
1) przedsiębiorcy zajmującemu się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi podlegającymi refundacji,
2) podmiotowi prowadzącemu aptekę, kierownikowi apteki lub osobie wydającej leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne objęte refundacją, w związku z realizacją recept na refundowane leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne
– sprzedaży uwarunkowanej, upustów, rabatów, bonifikat, pakietów i programów lojalnościowych, darowizn, nagród, wycieczek, gier losowych, zakładów wzajemnych, wszelkich form użyczeń, transakcji wiązanych, wszelkiego rodzaju talonów i bonów, a także udzielania innych niewymienionych z nazwy korzyści majątkowych lub osobistych dla świadczeniobiorców oraz osób uprawnionych.
4. Przepisy ust. 1–3 stosuje się do podmiotów realizujących zaopatrzenie na zlecenie w zakresie wyrobów medycznych oraz dostawców tych wyrobów.
5. Jeżeli w decyzji o objęciu refundacją leku, środka specjalnego przeznaczenia żywieniowego lub wyrobu medycznego, ustalano instrumenty dzielenia ryzyka, o których mowa w art. 11 ust. 2 pkt 7, które stanowią którąkolwiek z korzyści majątkowych lub osobistych, o których mowa w ust. 3, przepisów ust. 1–3 nie stosuje się w zakresie tych instrumentów.
Rozdział 7
Kary administracyjne
Art. 50. 1. Karze pieniężnej podlega, kto wbrew przepisom:
1) art. 6 stosuje inne odpłatności i dopłaty za leki, środki spożywcze specjalnego przeznaczenia żywieniowego lub wyroby medyczne;
2) art. 7 stosuje inne niż urzędowe marże hurtowe lub marże detaliczne na leki, środki spożywcze specjalnego przeznaczenia żywieniowego lub wyroby medyczne;
3) art. 8 stosuje inne niż ustalone w decyzji administracyjnej o objęciu refundacją ceny zbytu na leki, środki spożywcze specjalnego przeznaczenia żywieniowego lub wyroby medyczne;
4) art. 49 ust. 3 udziela którejkolwiek z korzyści majątkowych lub osobistych, o których mowa w tym przepisie.
2. Karę pieniężną, o której mowa w ust. 1 pkt 1–3, wymierza się w wysokości wartości sprzedanych z naruszeniem przepisów ustawy leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych powiększonej o wartość do 5% obrotu produktami, w stosunku do których wydana została decyzja administracyjna o objęciu refundacją, osiągniętego w poprzednim roku kalendarzowym.
3. Przy ustalaniu wysokości tej wartości bierze się pod uwagę:
1) cenę zbytu netto w przypadku wnioskodawcy,
2) cenę hurtową netto w przypadku podmiotu uprawnionego do obrotu hurtowego,
3) cenę detaliczną netto w przypadku podmiotu uprawnionego do obrotu detalicznego
– otrzymanej w poprzednim roku kalendarzowym kwoty z tytułu refundacji.
4. Karę pieniężną, o której mowa w ust. 1 pkt 4 wymierza się w wysokości do 5% wartości netto obrotu produktami, w stosunku do których wydana została decyzja administracyjna o objęciu refundacją osiągniętego w poprzednim roku kalendarzowym.
5. W przypadku, gdy podmiot ukarany nie wykazuje obrotu produktami, w stosunku do których wydana została decyzja administracyjna o objęciu refundacją, karę pieniężną, o której mowa w ust. 1 pkt 4, wymierza się w wysokości stukrotnej wartości udzielonej korzyści majątkowej lub osobistej, o której mowa w art. 49 ust. 3.
Art. 51. Karze pieniężnej podlega wnioskodawca, który nie dotrzymał określonych w decyzji administracyjnej o objęciu refundacją postanowień w zakresie instrumentów dzielenia ryzyka. Kara ta wymierzana jest w kwocie stanowiącej dwukrotność wartości poniesionych przez Fundusz kosztów refundacji związanych z niedotrzymaniem postanowień decyzji.
Art. 52. 1. Karze pieniężnej podlega ten, kto wbrew przepisowi art. 49 ust. 1:
1) uzależnia zawarcie umowy dotyczącej leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych podlegających refundacji lub uzależnia treść tej umowy, od przyjęcia lub spełnienia przez:
a) innego przedsiębiorcę zajmującego się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi podlegającymi refundacji,
b) podmiot prowadzący aptekę,
c) kierownika apteki,
d) osobę wydającą leki, środki spożywcze specjalnego przeznaczenia żywieniowego, wyroby medyczne objęte refundacją
– świadczenia niezwiązanego z przedmiotem tej umowy, w tym którejkolwiek korzyści majątkowej lub osobistej, o której mowa w art. 49 ust. 3;
2) stosuje wobec podmiotów wymienionych w pkt 1 niejednolite warunki umów.
2. Kara pieniężna, o której mowa w ust. 1, wymierzana jest w kwocie stanowiącej równowartość 3% wartości netto obrotu produktami, w stosunku do których wydana została decyzja administracyjna o objęciu refundacją, osiągniętego w poprzednim roku kalendarzowym.
Art. 52a.82) 1. Karze pieniężnej podlega osoba uprawniona, z wyłączeniem lekarza ubezpieczenia zdrowotnego, felczera ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, pielęgniarki ubezpieczenia zdrowotnego i położnej ubezpieczenia zdrowotnego, w rozumieniu ustawy o świadczeniach, o których mowa w art. 15a ust. 1 i 2 ustawy z dnia 15 lipca 2011 r. o zawodach pielęgniarki i położnej, która wystawiła receptę na refundowany lek, środek spożywczy specjalnego przeznaczenia żywieniowego albo wyrób medyczny, albo podmiot wykonujący działalność leczniczą, w ramach którego wystawiono receptę na refundowany lek, środek spożywczy specjalnego przeznaczenia żywieniowego albo wyrób medyczny albo zlecenie na wyrób medyczny, w przypadku:
1) uniemożliwienia czynności kontrolnych;
2) niewykonania w terminie zaleceń pokontrolnych.
2. Karę pieniężną w przypadku, o którym mowa w ust. 1:
1) pkt 1, wymierza się w kwocie stanowiącej równowartość,
2) pkt 2, wymierza się w kwocie do równowartości
– kwoty refundacji za okres objęty kontrolą.
3. Okres objęty kontrolą, o której mowa w ust. 2, nie może być dłuższy niż 5 lat, licząc od zakończenia roku kalendarzowego, w którym nastąpiła refundacja.
Art. 53. 1. Kary pieniężne, o których mowa w art. 50 ust. 1 pkt 1–3, art. 51 i art. 52, nakłada minister właściwy do spraw zdrowia w drodze decyzji administracyjnej.
2. Karę pieniężną, o której mowa w art. 50 ust. 1 pkt 4, nakłada w drodze decyzji administracyjnej wojewódzki inspektor farmaceutyczny, na którego obszarze działania doszło do naruszenia przepisów art. 49 ust. 3.
2a.83) Karę pieniężną, o której mowa w art. 52a, nakłada w drodze decyzji administracyjnej dyrektor oddziału wojewódzkiego Funduszu. Od decyzji przysługuje odwołanie do Prezesa Funduszu.
3.84) Przy ustalaniu wysokości kar pieniężnych, o których mowa w ust. 1, 2 oraz art. 52a, należy uwzględnić w szczególności okres, stopień oraz okoliczności naruszenia przepisów ustawy, a także uprzednie naruszenia przepisów ustawy.
4.84) Kary pieniężne stanowią przychód Funduszu. Prezes Funduszu jest wierzycielem w rozumieniu ustawy z dnia 17 czerwca 1966 r. o postępowaniu egzekucyjnym w administracji (Dz. U. z 2016 r. poz. 599 i 868). Zażalenia na postanowienia Prezesa Funduszu rozpatruje minister właściwy do spraw zdrowia.
5.85) Karę pieniężną uiszcza się w terminie 7 dni od dnia, w którym decyzja stała się ostateczna na rachunek bankowy wskazany przez Prezesa Funduszu. Od kary pieniężnej nieuiszczonej w terminie nalicza się odsetki ustawowe za opóźnienie.
6.85) Egzekucja kary pieniężnej wraz z odsetkami, o których mowa w ust. 5, następuje w trybie przepisów o postępowaniu egzekucyjnym w administracji.
7.86) Kara pieniężna, o której mowa w ust. 1, 2 i 2a, ulega przedawnieniu z upływem 5 lat, licząc od dnia, w którym decyzja ustalająca karę stała się ostateczna.
Rozdział 8
Przepisy karne
Art. 54. 1. Kto, zajmując się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego lub wyrobami medycznymi podlegającymi refundacji ze środków publicznych, przyjmuje korzyść majątkową lub osobistą albo jej obietnicę lub takiej korzyści żąda w zamian za zachowanie wywierające wpływ na:
1) poziom obrotu lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego lub wyrobami medycznymi podlegającymi refundacji ze środków publicznych;
2) obrót lub powstrzymanie się od obrotu konkretnym lekiem, środkiem spożywczym specjalnego przeznaczenia żywieniowego lub wyrobem medycznym podlegającym refundacji ze środków publicznych;
– podlega karze pozbawienia wolności od 6 miesięcy do 8 lat.
2. Tej samej karze podlega, kto będąc osobą uprawnioną do wystawiania recept na leki, środki spożywcze specjalnego przeznaczenia żywieniowego lub wyroby medyczne, podlegające refundacji ze środków publicznych lub zleceń, o których mowa w art. 38 ust. 1, żąda lub przyjmuje korzyść majątkową lub osobistą albo jej obietnicę w zamian za wystawienie recepty lub zlecenia lub powstrzymanie się od ich wystawienia.
3. Tej samej karze podlega, kto będąc osobą zaopatrującą świadczeniodawcę w leki, środki spożywcze specjalnego przeznaczenia żywieniowego lub wyroby medyczne albo będąc świadczeniodawcą lub osobą reprezentującą świadczeniodawcę żąda lub przyjmuje korzyść majątkową lub osobistą, w zamian za zakup leku, środka spożywczego specjalnego przeznaczenia żywieniowego lub wyrobu medycznego podlegającego refundacji ze środków publicznych.
4. Tej samej karze podlega, kto w przypadkach określonych w ust. 1–3 udziela lub obiecuje udzielić korzyści majątkowej lub osobistej.
5. W wypadku mniejszej wagi sprawca czynu określonego w ust. 1–4
– podlega karze pozbawienia wolności do lat 3.
6. Nie podlega karze sprawca przestępstwa określonego w ust. 4, jeżeli korzyść majątkowa lub osobista albo jej obietnica zostały przyjęte, a sprawca zawiadomił o tym fakcie organ powołany do ścigania przestępstw i ujawnił wszystkie istotne okoliczności przestępstwa, zanim organ ten o nim się dowiedział.
Art. 55. Odpis prawomocnego wyroku skazującego osobę uprawnioną do wystawiania recept na refundowane leki, środki spożywcze specjalnego przeznaczenia żywieniowego lub wyroby medyczne, za przestępstwo określone w art. 54 ustawy lub art. 228–230, art. 286 lub art. 296a ustawy z dnia 6 czerwca 1997 r. – Kodeks karny, sąd przesyła podmiotowi zobowiązanemu do finansowania świadczeń ze środków publicznych.
Rozdział 9
Zmiany w przepisach obowiązujących, przepisy przejściowe i końcowe
Art. 56–66. (pominięte)
Art. 67. 1. Minister właściwy do spraw zdrowia, w terminie miesiąca od dnia wejścia w życie niniejszego przepisu, wzywa podmiot odpowiedzialny, importera równoległego, w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, a także wytwórcę wyrobu medycznego lub jego autoryzowanego przedstawiciela, dystrybutora lub importera, w rozumieniu ustawy z dnia 20 maja 2010 r. o wyrobach medycznych, którego lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny jest objęty wykazami:
1) wydanymi na podstawie art. 31d ustawy, o której mowa w art. 63, dotyczącym świadczeń gwarantowanych z zakresu:
a) programów zdrowotnych w części dotyczącej terapeutycznych programów zdrowotnych, z zastrzeżeniem art. 70,
b) leczenia szpitalnego oraz ambulatoryjnej opieki specjalistycznej w części dotyczącej leków stosowanych w chemioterapii,
2) o których mowa w art. 36 ust. 5, art. 37 ust. 2 i art. 38 ust. 6 tej ustawy
– do przeprowadzenia negocjacji w zakresie ustalenia urzędowej ceny zbytu oraz instrumentów dzielenia ryzyka, o których mowa w art. 11 ust. 5.
2. Wezwanie, o którym mowa w ust. 1, zawiera żądanie przedstawienia informacji i zobowiązań, o których mowa w art. 24 ust. 2, art. 25 pkt 4 i pkt 6 lit. e oraz art. 26 pkt 1 lit. a–f i j–l. Podmioty wskazane w ust. 1 są obowiązane przedstawić te informacje w terminie 60 dni od dnia doręczenia wezwania.
3. Podmiot, o którym mowa w ust. 1 lub w przypadku przedłożenia odpowiedniej umowy, o której mowa w art. 24 ust. 2 pkt 5, jego przedstawiciel, staje się z mocy prawa wnioskodawcą w rozumieniu niniejszej ustawy, z chwilą przekazania ministrowi właściwemu do spraw zdrowia informacji, o których mowa w ust. 2.
4. W przypadku stwierdzenia, że przedłożone przez wnioskodawcę informacje, o których mowa w ust. 2 są niepełne, minister właściwy do spraw zdrowia wzywa go do ich uzupełnienia w terminie 7 dni.
5. W przypadku nieprzedłożenia informacji, o których mowa w ust. 2 lub niedokonania ich uzupełnienia w terminie, o którym mowa w ust. 3, lek, środek spożywczy specjalnego przeznaczenia żywieniowego, wyrób medyczny nie zostaje umieszczony w pierwszym wykazie, o którym mowa w art. 68.
6. Informacje, o których mowa w ust. 2, w zakresie określonym w art. 26 pkt 1 lit. e–f dotyczą okresu od 1 maja 2010 r. do 30 kwietnia 2011 r.
7. Negocjacje w sprawie ustalenia urzędowej ceny zbytu dotyczą proponowanej w informacji przedstawionej przez wnioskodawcę wskazanego w ust. 3 ceny zbytu netto.
8. Urzędowa cena zbytu ustalona w wyniku negocjacji nie może być wyższa niż maksymalna cena detaliczna brutto leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego określona w wykazie, o którym mowa w art. 36 ust. 5, art. 37 ust. 2 i art. 38 ust. 6 ustawy, o której mowa w art. 63, w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy, pomniejszona o marżę hurtową w wysokości 8,91% liczoną od ceny urzędowej hurtowej oraz marżę detaliczną w wysokości:
| Cena hurtowa w złotych | Marża detaliczna liczona od ceny hurtowej |
| 0–3,60 | 40% |
| 3,61–4,80 | 1,44 zł |
| 4,81–6,50 | 30% |
| 6,51–9,75 | 1,95 zł |
| 9,76–14,00 | 20% |
| 14,01–15,55 | 2,80 zł |
| 15,56–30,00 | 18% |
| 30,01–33,75 | 5,40 zł |
| 33,76–50,00 | 16% |
| 50,01–66,67 | 8,00 zł |
| 66,68–100,00 | 12% |
| powyżej 100,00 | 12,00 zł |
9. Negocjacje przeprowadza Komisja. Do negocjacji stosuje się art. 18 ust. 3, art. 19 ust. 1 i 2 pkt 2–7, art. 20 i art. 22 oraz przepisy wykonawcze wydane na podstawie art. 23.
10. Minister właściwy do spraw zdrowia wydaje decyzję, o której mowa w art. 11 ust. 1, uwzględniając:
1) stanowisko Komisji,
2) konkurencyjność cenową
– biorąc pod uwagę równoważenie interesów świadczeniobiorców i przedsiębiorców zajmujących się wytwarzaniem lub obrotem lekami, środkami spożywczymi specjalnego przeznaczenia żywieniowego, wyrobami medycznymi, możliwości płatnicze podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych oraz działalność naukowo-badawczą i inwestycyjną wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA).
11. Decyzja, o której mowa w ust. 10, zawiera elementy, o których mowa w art. 11 ust. 2 pkt 1–8.
12. Decyzje, o których mowa w ust. 10, wydaje się na okres 2 lat.
Art. 68. 1. Minister właściwy do spraw zdrowia ustali pierwszy wykaz, o którym mowa w art. 37 ust. 1 na dzień 1 stycznia 2012 r.
2. Pierwszy wykaz zawiera dane, o których mowa w art. 37 ust. 2, przy czym dane, o których mowa w art. 37 ust. 2 pkt 2–7, są ustalone w sposób określony w ust. 3.
3. Lekom, środkom spożywczym specjalnego przeznaczenia żywieniowego, wyrobom medycznym objętym pierwszym wykazem:
1) nadaje się kategorię dostępności refundacyjnej, o której mowa w art. 6 ust. 1:
a) pkt 1 lit. a – w przypadku, o którym mowa w ust. 1 pkt 1,
b) pkt 1 lit. b – w przypadku, o którym mowa w ust. 1 pkt 2,
c) pkt 2 – w przypadku, o którym mowa w ust. 1 pkt 3,
d) pkt 3 – w przypadku, o którym mowa w ust. 1 pkt 4;
2) przypisuje się limity finansowania i ustala grupy limitowe, na zasadach określonych w art. 15;
3) przypisuje się odpłatności na zasadach określonych w art. 14;
4) ustala się urzędową cenę zbytu w trybie, o którym mowa w art. 67.
Art. 69. Umowy o udzielanie świadczeń opieki zdrowotnej z zakresu programów zdrowotnych w części dotyczącej terapeutycznych programów zdrowotnych oraz z zakresu leczenia szpitalnego i ambulatoryjnej opieki specjalistycznej w części dotyczącej leków stosowanych w chemioterapii, zawarte przed dniem wejścia w życie niniejszej ustawy, są realizowane po tym dniu na zasadach dotychczasowych, nie dłużej jednak niż do dnia 30 czerwca 2012 r.
Art. 70. 1. Świadczenia chemioterapii niestandardowej określone w przepisach wydanych na podstawie art. 31d ustawy, o której mowa w art. 63 w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy, realizowane przed tym dniem, są realizowane na dotychczasowych zasadach nie dłużej jednak niż do dnia 31 grudnia 2014 r.
2. W okresie 3 lat od dnia wejścia w życie niniejszej ustawy mogą być kierowane do dyrektora oddziału wojewódzkiego Narodowego Funduszu Zdrowia wnioski świadczeniodawcy dotyczące rozpoczęcia realizacji świadczeń chemioterapii niestandardowej określonych w przepisach wydanych na podstawie art. 31d ustawy, o której mowa w art. 63 w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy. Wnioski te są rozpatrywane zgodnie z dotychczasowymi zasadami. Świadczenia chemioterapii niestandardowej realizowane na podstawie tych wniosków są realizowane przez okres określony w przepisach wydanych na podstawie art. 31d ustawy, o której mowa w art. 63 w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy.
3. Do kontynuacji u świadczeniobiorcy chemioterapii niestandardowej danym lekiem w danym wskazaniu, stosuje się ust. 2 zdanie drugie i trzecie.
4. Świadczenia chemioterapii niestandardowej określone w przepisach wydanych na podstawie art. 31d ustawy, o której mowa w art. 63 w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy nie mogą być realizowane przy wykorzystaniu leku, który został dopuszczony do obrotu, zgodnie z przepisami ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, po dniu 31 grudnia 2011 r.
5. Jeżeli podmiot odpowiedzialny złoży wniosek, o którym mowa w art. 24 ust. 1 pkt 1, dotyczący leku stosowanego w świadczeniu, o którym mowa w ust. 1, w zakresie wskazań do stosowania zgodnych z określonymi w Charakterystyce Produktu Leczniczego w rozumieniu ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, w terminie do dnia 31 grudnia 2012 r., opłat, o których mowa w art. 32 i art. 35 ust. 3 nie pobiera się.
Art. 71. 1. Wnioski złożone po dniu 30 czerwca 2011 r. na podstawie art. 39 ustawy, o której mowa w art. 63, w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy, uzupełnia się w terminie 14 dni od dnia doręczenia wezwania ministra właściwego do spraw zdrowia, o informacje, zgodnie z art. 67 ust. 2.
2. Do wniosków tych przepis art. 67 ust. 7–12 stosuje się odpowiednio.
3. Jeżeli wniosek, o którym mowa w ust. 1 dotyczy leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, nieposiadającego swojego odpowiednika w danym wskazaniu na wykazach, o których mowa w art. 36 ust. 5, art. 37 ust. 2 i art. 38 ust. 6 ustawy, o której mowa w art. 63, w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy, przepis art. 31i, art. 39 ust. 2d, 2e pkt 1 oraz ust. 2f–2j ustawy, o której mowa w art. 63, w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy, stosuje się.
Art. 72. Leki zawarte w wykazie leków podstawowych, o którym mowa w art. 36 ust. 5 pkt 1, oraz leki, środki spożywcze specjalnego przeznaczenia żywieniowego, zawarte w wykazie, o którym mowa w art. 37 ust. 2 pkt 2 ustawy, o której mowa w art. 63, w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy wydawane za odpłatnością ryczałtową, minister właściwy do spraw zdrowia kwalifikuje do odpłatności ryczałtowej, o ile zgodnie z aktualną wiedzą medyczną stosuje się je dłużej niż 30 dni.
Art. 73. W celu obliczenia po raz pierwszy kwoty przekroczenia, o której mowa w art. 4 ust. 1, za planową kwotę refundacji w grupie limitowej w roku 2011 rozumie się wykonaną kwotę refundacji w tej grupie w roku 2011.
Art. 74. 1. Wysokość całkowitego budżetu na refundację w 2012 r. jest równa kwocie kosztów poniesionych w 2010 r. na finansowanie świadczeń gwarantowanych określonych w przepisach wydanych na podstawie:
1) art. 31d ustawy, o której mowa w art. 63, w zakresie dotyczącym:
a) programów zdrowotnych w części dotyczącej leków stosowanych w terapeutycznych programach zdrowotnych,
b) leczenia szpitalnego oraz ambulatoryjnej opieki specjalistycznej w części dotyczącej leków stosowanych w chemioterapii;
2) art. 36 ust. 4 i 5, art. 37 ust. 2 i art. 38 ust. 6 ustawy, o której mowa w art. 63.
2. W przypadku gdy po zatwierdzeniu sprawozdania finansowego Funduszu za 2011 r. koszty poniesione na finansowanie świadczeń gwarantowanych w 2011 r., o których mowa w ust. 1, będą inne niż ujęte w planie finansowym Funduszu na 2012 r. Prezes Funduszu dokonuje zmiany planu finansowego. Wysokość całkowitego budżetu na refundację w 2012 r. Prezes Funduszu dostosuje do wysokości kosztów poniesionych na finansowanie świadczeń gwarantowanych, o których mowa w ust. 1, w 2011 r.
3. Wysokość całkowitego budżetu na refundację w latach 2013 i 2014 jest równa kwocie poniesionych przez Fundusz wydatków związanych z finansowaniem świadczeń gwarantowanych, o których mowa w art. 15 ust. 2 pkt 14–18 ustawy, o której mowa w art. 63, w brzmieniu nadanym niniejszą ustawą, w roku 2011, a począwszy od roku 2015 nie może być niższa od tej kwoty.
4. Plany finansowe Funduszu począwszy od roku 2013 są ustalane z uwzględnieniem ust. 3.
Art. 75. Urzędowa marża hurtowa, o której mowa w art. 7 ust. 1–3:
1) w roku 2012 – wynosi 7% urzędowej ceny zbytu;
2) w roku 2013 – wynosi 6% urzędowej ceny zbytu.
Art. 76. 1. Do rozpatrywania wniosków, o których mowa w art. 39 ustawy, o której mowa w art. 63, w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy, złożonych przed dniem wejścia w życie niniejszej ustawy, stosuje się przepisy niniejszej ustawy. Opłat, o których mowa w art. 32 ust. 1 i art. 35 ust. 3 nie pobiera się.
2. Dla wniosków, o których mowa w ust. 1, bieg terminów, o których mowa w art. 31, liczy się od dnia wejścia w życie niniejszej ustawy.
Art. 77. 1. Zlecenia ministra właściwego do spraw zdrowia, o których mowa w art. 31e ustawy, o której mowa w art. 63, dotyczące świadczeń gwarantowanych, o których mowa w art. 15 ust. 2:
1) pkt 2 i 3 ustawy, o której mowa w art. 63, z zakresu leczenia szpitalnego oraz ambulatoryjnej opieki specjalistycznej w części dotyczącej leków stosowanych w chemioterapii,
2) pkt 13 ustawy, o której mowa w art. 63, z zakresu programów zdrowotnych w części dotyczącej terapeutycznych programów zdrowotnych,
3) pkt 14 ustawy, o której mowa w art. 63, w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy
– skierowane do Prezesa Agencji Oceny Technologii Medycznych przed dniem wejścia w życie niniejszej ustawy zachowują ważność i są rozpatrywane na zasadach dotychczasowych.
2. Rekomendacja Prezesa Agencji Oceny Technologii Medycznych wydana na podstawie zlecenia, o którym mowa w ust. 1, stanowi jedną z przesłanek uchylenia decyzji administracyjnej o objęciu refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego oraz wyrobu medycznego.
Art. 78. Rada Konsultacyjna, o której mowa w art. 31s ustawy, o której mowa w art. 63, w brzmieniu obowiązującym przed dniem wejścia życie niniejszej ustawy, działa na podstawie dotychczasowych przepisów i wykonuje zadania Rady Przejrzystości, o której mowa w art. 31s ustawy, o której mowa w art. 63, w brzmieniu nadanym niniejszą ustawą, do czasu jej powołania, nie dłużej jednak niż przez 3 miesiące od dnia wejścia w życie niniejszej ustawy.
Art. 79. 1. Zespół do spraw Gospodarki Lekami, o którym mowa w art. 7 ustawy, o której mowa w art. 59, działa na podstawie dotychczasowych przepisów i wykonuje zadania Komisji Ekonomicznej, o której mowa w art. 17, do czasu jej powołania, nie dłużej jednak niż przez miesiąc od dnia wejścia w życie niniejszej ustawy.
2. Członkowie Zespołu do spraw Gospodarki Lekami, o którym mowa w art. 7 ustawy, o której mowa w art. 59, składają oświadczenia, o których mowa w art. 17, przed pierwszym posiedzeniem Zespołu, jednakże nie później niż w terminie 14 dni od dnia wejścia w życie niniejszej ustawy.
Art. 80. 1. Fundusz podejmie działania konieczne do zawarcia i niezwłocznie zawiera umowy, o których mowa w art. 41 i art. 48, z podmiotami prowadzącymi apteki oraz osobami uprawnionymi, o których mowa w art. 2 pkt 14 lit. b i c.
2. Strony umów określonych w art. 34 ust. 2 ustawy, o której mowa w art. 63, dostosują ich treść do przepisów niniejszej ustawy w terminie do dnia 30 czerwca 2012 r.
3. Fundusz w terminie 15 dni od dnia wejścia w życie niniejszego przepisu:
1) przekaże ministrowi właściwemu do spraw zdrowia zestawienie, o którym mowa w art. 102 ust. 5 pkt 26 ustawy, o której mowa w art. 63, w brzmieniu nadanym niniejszą ustawą,
2) poda do publicznej wiadomości informacje, o których mowa w art. 102 ust. 5 pkt 27 i 29 ustawy, o której mowa w art. 63, w brzmieniu nadanym niniejszą ustawą
– za okres od dnia 1 stycznia 2011 r. do dnia ogłoszenia niniejszej ustawy.
4. Minister właściwy do spraw zdrowia podejmie czynności mające na celu powołanie Komisji Ekonomicznej, o której mowa w art. 17, oraz Rady Przejrzystości, o której mowa w art. 31s ustawy, o której mowa w art. 63, w brzmieniu nadanym niniejszą ustawą.
Art. 81. Recepty wystawione przed dniem wejścia w życie niniejszej ustawy są realizowane na dotychczasowych zasadach.
Art. 82. Do kontroli wszczętych i niezakończonych przed dniem wejścia w życie niniejszej ustawy stosuje się przepisy dotychczasowe.
Art. 83. Objęcie pierwszym wykazem, o którym mowa w art. 68, produktów, które w dniu wejścia w życie niniejszej ustawy dopuszczone były do obrotu jako produkty lecznicze zgodnie z przepisami ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, na podstawie dokumentacji, o której mowa w art. 17 ust. 3 ustawy, o której mowa w art. 60, w brzmieniu obowiązującym przed dniem wejścia w życie niniejszej ustawy, jest równoznaczne z powiadomieniem, o którym mowa w art. 29 ustawy z dnia 25 sierpnia 2006 r. o bezpieczeństwie żywności i żywienia.
Art. 84. Rada Ministrów po upływie dwóch lat od dnia wejścia w życie niniejszej ustawy przedłoży Sejmowi Rzeczypospolitej Polskiej sprawozdanie z wykonania tej ustawy wraz z oceną skutków jej stosowania.
Art. 85. 1. Dotychczasowe przepisy wykonawcze, wydane na podstawie:
1) art. 45 ust. 3 ustawy, o której mowa w art. 58,
2) art. 31d ustawy, o której mowa w art. 63 utrzymane w mocy na podstawie art. 3 ust. 1 ustawy z dnia 23 lipca 2010 r. o zmianie ustawy o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych (Dz. U. poz. 1116),
3) art. 31j, art. 31s ust. 13, art. 36 ust. 5 pkt 4 i 5, art. 39 ust. 9, art. 39c, art. 159 ust. 5 i art. 190 ust. 2 ustawy, o której mowa w art. 63
– zachowują moc do dnia wejścia w życie przepisów wykonawczych wydanych na podstawie art. 45 ust. 5 ustawy, o której mowa w art. 58, w brzmieniu nadanym niniejszą ustawą, art. 31d, 31j, 31s ust. 25 i art. 159 ust. 5 ustawy, o której mowa w art. 63, w brzmieniu nadanym niniejszą ustawą, oraz art. 6 ust. 10, art. 24 ust. 7, art. 35 ust. 10 i art. 38 ust. 4, jednak nie dłużej niż przez okres 24 miesięcy od dnia wejścia w życie niniejszej ustawy.
2. Zachowane w mocy akty wykonawcze, o których mowa w ust. 1, wydane na podstawie art. 31d ustawy, o której mowa w art. 63, mogą być zmienione przez ministra właściwego do spraw zdrowia, w drodze rozporządzenia, w granicach określonych w art. 31d ustawy, o której mowa w art. 63, w brzmieniu nadanym niniejszą ustawą.
Art. 86. Ustawa wchodzi w życie z dniem 1 stycznia 2012 r., z wyjątkiem:
1) art. 11, art. 17–23, art. 41 ust. 1–5 i 8, art. 48 ust. 1–6, art. 63 pkt 26, art. 67, art. 68 ust. 2 i 3, art. 71, art. 74, art. 79 i art. 80, które wchodzą w życie po upływie 14 dni od dnia ogłoszenia87);
2) art. 63 pkt 13, który wchodzi w życie z dniem 1 lipca 2012 r.
1) Niniejsza ustawa dokonuje w zakresie swojej regulacji wdrożenia dyrektywy Rady 89/105/EWG z dnia 21 grudnia 1988 r. dotyczącej przejrzystości środków regulujących ustalanie cen na produkty lecznicze przeznaczone do użytku przez człowieka oraz włączenia ich w zakres krajowego systemu ubezpieczeń zdrowotnych (Dz. Urz. WE L 40 z 11.02.1989, str. 8; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 5, t. 1, str. 345).
2) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2015 r. poz. 1240, 1269, 1365, 1569, 1692, 1735, 1830, 1844, 1893, 1916, 1991 i 1994 oraz z 2016 r. poz. 65, 652 i 960.
3) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2008 r. poz. 1505 i 1570, z 2009 r. poz. 97, 206, 753, 788 i 817, z 2010 r. poz. 513 i 679, z 2011 r. poz. 322, 451, 622, 654, 657 i 696, z 2012 r. poz. 1342 i 1544, z 2013 r. poz. 1245, z 2014 r. poz. 822 i 1491, z 2015 r. poz. 28, 277, 788, 875, 1771, 1830, 1918, 1926 i 1991 oraz z 2016 r. poz. 823 i 960.
4) W brzmieniu ustalonym przez art. 13 pkt 1 ustawy z dnia 9 października 2015 r. o zmianie ustawy o systemie informacji w ochronie zdrowia oraz niektórych innych ustaw (Dz. U. poz. 1991), która weszła w życie z dniem 12 grudnia 2015 r.
5) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2015 r. poz. 699, 875, 978, 1197, 1268, 1272, 1618, 1649, 1688, 1712, 1844 i 1893 oraz z 2016 r. poz. 65, 352, 615, 780, 868, 903 i 960.
6) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 7.
7) W brzmieniu ustalonym przez art. 13 pkt 2 ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
8) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2015 r. poz. 788, 855, 1066, 1918, 1991, 1994 i 2281.
9) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 10.
10) W brzmieniu ustalonym przez art. 13 pkt 3 lit. a ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
11) Dodany przez art. 13 pkt 3 lit. b ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
12) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 13.
13) W brzmieniu ustalonym przez art. 13 pkt 4 lit. a ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
14) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 15.
15) W brzmieniu ustalonym przez art. 13 pkt 4 lit. b ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
16) Dodany przez art. 13 pkt 4 lit. c ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
17) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 18.
18) Przez art. 13 pkt 4 lit. d ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
19) W brzmieniu ustalonym przez art. 2 pkt 1 ustawy z dnia 18 marca 2016 r. o zmianie ustawy o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych oraz niektórych innych ustaw (Dz. U. poz. 652), która weszła w życie z dniem 12 czerwca 2016 r.
20) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 21.
21) W brzmieniu ustalonym przez art. 13 pkt 5 lit. a ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
22) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 23.
23) W brzmieniu ustalonym przez art. 13 pkt 5 lit. b ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
24) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 25.
25) W brzmieniu ustalonym przez art. 13 pkt 5 lit. c ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
26) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 27.
27) Przez art. 13 pkt 5 lit. d ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
28) Dodana przez art. 13 pkt 6 lit. a tiret pierwsze ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
29) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 30.
30) W brzmieniu ustalonym przez art. 13 pkt 6 lit. a tiret drugie ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
31) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 32.
32) W brzmieniu ustalonym przez art. 13 pkt 6 lit. a tiret trzecie ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
33) Dodana przez art. 13 pkt 6 lit. b tiret pierwsze ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
34) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 35.
35) W brzmieniu ustalonym przez art. 13 pkt 6 lit. b tiret drugie ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
36) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 37.
37) W brzmieniu ustalonym przez art. 13 pkt 6 lit. b tiret trzecie ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
38) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 39.
39) W brzmieniu ustalonym przez art. 13 pkt 7 lit. a tiret pierwsze ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
40) Dodana przez art. 13 pkt 7 lit. a tiret drugie ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
41) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 42.
42) Przez art. 13 pkt 7 lit. b ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
43) Dodany przez art. 13 pkt 8 ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
44) Wprowadzenie do wyliczenia w tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 45.
45) Wprowadzenie do wyliczenia w brzmieniu ustalonym przez art. 13 pkt 9 lit. a ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
46) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 47.
47) W brzmieniu ustalonym przez art. 13 pkt 9 lit. b ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
48) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 49.
49) W brzmieniu ustalonym przez art. 13 pkt 9 lit. c ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
50) W tym brzmieniu obowiązuje do wejścia w życie zmiany, o której mowa w odnośniku 51.
51) W brzmieniu ustalonym przez art. 13 pkt 10 ustawy, o której mowa w odnośniku 4; wejdzie w życie z dniem 1 stycznia 2017 r.
52) Dodany przez art. 2 pkt 2 ustawy, o której mowa w odnośniku 19.
53) Dodany przez art. 13 pkt 11 ustawy, o której mowa w odnośniku 4.
54) W brzmieniu ustalonym przez art. 13 pkt 12 lit. a ustawy, o której mowa w odnośniku 4.
55) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2014 r. poz. 1491 i 1877, z 2015 r. poz. 978, 1640, 1893 i 1991 oraz z 2016 r. poz. 65 i 652.
56) Dodany przez art. 13 pkt 12 lit. b ustawy, o której mowa w odnośniku 4.
57) W brzmieniu ustalonym przez art. 13 pkt 12 lit. c ustawy, o której mowa w odnośniku 4.
58) Dodany przez art. 13 pkt 12 lit. d ustawy, o której mowa w odnośniku 4.
59) W brzmieniu ustalonym przez art. 13 pkt 12 lit. e ustawy, o której mowa w odnośniku 4.
60) Dodany przez art. 13 pkt 12 lit. f ustawy, o której mowa w odnośniku 4.
61) W brzmieniu ustalonym przez art. 13 pkt 12 lit. g ustawy, o której mowa w odnośniku 4.
62) Dodany przez art. 13 pkt 13 ustawy, o której mowa w odnośniku 4.
63) Dodany przez art. 13 pkt 14 ustawy, o której mowa w odnośniku 4.
64) W brzmieniu ustalonym przez art. 44 pkt 1 ustawy z dnia 9 października 2015 r. o zmianie ustawy o terminach zapłaty w transakcjach handlowych, ustawy – Kodeks cywilny oraz niektórych innych ustaw (Dz. U. poz. 1830), która weszła w życie z dniem 1 stycznia 2016 r.
65) W brzmieniu ustalonym przez art. 2 pkt 3 lit. a ustawy, o której mowa w odnośniku 19.
66) Dodany przez art. 2 pkt 3 lit. b ustawy, o której mowa w odnośniku 19.
67) W brzmieniu ustalonym przez art. 2 pkt 3 lit. c ustawy, o której mowa w odnośniku 19.
68) Dodany przez art. 2 pkt 4 ustawy, o której mowa w odnośniku 19.
69) W brzmieniu ustalonym przez art. 2 pkt 5 ustawy, o której mowa w odnośniku 19.
70) W brzmieniu ustalonym przez art. 44 pkt 2 ustawy, o której mowa w odnośniku 64.
71) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2015 r. poz. 1633, 1893, 1991 i 2199 oraz z 2016 r. poz. 65, 960 i 1070.
72) Dodany przez art. 13 pkt 16 ustawy, o której mowa w odnośniku 4.
73) W brzmieniu ustalonym przez art. 13 pkt 17 lit. a ustawy, o której mowa w odnośniku 4.
74) Przez art. 13 pkt 17 lit. b ustawy, o której mowa w odnośniku 4.
75) Dodany przez art. 13 pkt 17 lit. c ustawy, o której mowa w odnośniku 4.
76) Przez art. 13 pkt 17 lit. d ustawy, o której mowa w odnośniku 4.
77) W brzmieniu ustalonym przez art. 13 pkt 17 lit. e ustawy, o której mowa w odnośniku 4.
78) Przez art. 13 pkt 17 lit. f ustawy, o której mowa w odnośniku 4.
79) W brzmieniu ustalonym przez art. 13 pkt 17 lit. g ustawy, o której mowa w odnośniku 4.
80) Dodany przez art. 13 pkt 17 lit. h ustawy, o której mowa w odnośniku 4.
81) Przez art. 13 pkt 17 lit. i ustawy, o której mowa w odnośniku 4.
82) Dodany przez art. 13 pkt 18 ustawy, o której mowa w odnośniku 4.
83) Dodany przez art. 13 pkt 19 lit. a ustawy, o której mowa w odnośniku 4.
84) W brzmieniu ustalonym przez art. 13 pkt 19 lit. b ustawy, o której mowa w odnośniku 4.
85) W brzmieniu ustalonym przez art. 44 pkt 3 ustawy, o której mowa w odnośniku 64.
86) Dodany przez art. 13 pkt 19 lit. c ustawy, o której mowa w odnośniku 4.
87) Ustawa została ogłoszona w dniu 13 czerwca 2011 r.
- Data ogłoszenia: 2016-09-23
- Data wejścia w życie: 2016-09-23
- Data obowiązywania: 2017-07-23
- Dokument traci ważność: 2017-10-04

- USTAWA z dnia 9 października 2015 r. o zmianie ustawy o systemie informacji w ochronie zdrowia oraz niektórych innych ustaw
- USTAWA z dnia 5 września 2016 r. o usługach zaufania oraz identyfikacji elektronicznej
- USTAWA z dnia 25 maja 2017 r. o zmianie ustawy o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych oraz niektórych innych ustaw
- USTAWA z dnia 7 lipca 2017 r. o zmianie ustawy o przeciwdziałaniu narkomanii oraz ustawy o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych
REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA