REKLAMA
Dziennik Ustaw - rok 2013 poz. 5
ROZPORZĄDZENIE
MINISTRA ZDROWIA1)
z dnia 11 grudnia 2012 r.
w sprawie leczenia krwią w podmiotach leczniczych wykonujących działalność leczniczą w rodzaju stacjonarne i całodobowe świadczenia zdrowotne, w których przebywają pacjenci ze wskazaniami do leczenia krwią i jej składnikami2)
Na podstawie art. 21 ust. 3 ustawy z dnia 22 sierpnia 1997 r. o publicznej służbie krwi (Dz. U. Nr 106, poz. 681, z późn. zm.3)) zarządza się, co następuje:
Rozdział 1
Przepisy ogólne
§ 1. [Zakres regulacji]
1) zadania kierownika podmiotu leczniczego, ordynatora oddziału lub innej osoby kierującej jednostką lub komórką organizacyjną przedsiębiorstwa podmiotu leczniczego, lekarzy i pielęgniarek (położnych);
2) sposób powoływania banku krwi w podmiocie leczniczym oraz jego lokalizację i organizację pracy;
3) sposób sprawowania nadzoru nad działaniem banku krwi w podmiocie leczniczym oraz nad stosowaniem leczenia krwią i jej składnikami w tym podmiocie.
1) niezwłoczne, całodobowe zaopatrzenie jednostek lub komórek organizacyjnych przedsiębiorstwa podmiotu leczniczego w krew i jej składniki;
2) badania z zakresu immunologii transfuzjologicznej warunkujące bezpieczne przetaczanie krwi i jej składników, zwane dalej „przetoczeniem”;
3) identyfikację i rejestrowanie wszelkich nieprzewidzianych zdarzeń związanych z przetoczeniem, a w szczególności błędów i wypadków, oraz sporządzanie raportów o tych zdarzeniach.
1) ustalenie wskazań do przetoczenia;
2) identyfikację biorcy, na podstawie danych, o których mowa w § 11 ust. 1 pkt 1, i kontrolę dokumentacji medycznej przed przetoczeniem;
3) zabieg przetoczenia;
4) prawidłowe udokumentowanie zabiegu przetoczenia;
5) sporządzanie raportów o zdarzeniach, o których mowa w § 2 pkt 3.
2. Pielęgniarka (położna) jest odpowiedzialna za:
1) czynności związane z pobieraniem próbek krwi od pacjentów;
2) identyfikację biorcy, na podstawie danych, o których mowa w § 11 ust. 1 pkt 1, i kontrolę dokumentacji medycznej przed przetoczeniem – na zlecenie lekarza;
3) obserwację biorcy w trakcie przetoczenia i niezwłoczne informowanie lekarza o objawach występujących w trakcie przetoczenia i po przetoczeniu mogących świadczyć o powikłaniu poprzetoczeniowym oraz o zdarzeniach, o których mowa w § 2 pkt 3;
4) wydawanie wypełnionego i podpisanego przez lekarza zamówienia na krew i jej składniki;
5) prawidłowe udokumentowanie zabiegu przetoczenia.
3. Naczelna pielęgniarka (położna) lub osoba nadzorująca pracę pielęgniarek (położnych), w porozumieniu z ordynatorem lub inną osobą kierującą jednostką lub komórką organizacyjną przedsiębiorstwa podmiotu leczniczego, ustala imienną listę pielęgniarek (położnych) uprawnionych do dokonywania przetoczeń i czynności związanych z tym zabiegiem, posiadających zaświadczenie o odbyciu szkolenia określone w przepisach wydanych na podstawie art. 21 ust. 2 ustawy z dnia 22 sierpnia 1997 r. o publicznej służbie krwi, zwanej dalej „ustawą”.
1) wypełnienie zamówienia na krew i jej składniki – dotyczy wyłącznie lekarza;
2) złożenie zamówienia na krew i jej składniki;
3) pobranie od pacjenta próbek krwi w celu wykonania badania grupy krwi i próby zgodności krwi dawcy i biorcy przed przetoczeniem, zwanego dalej „próbą zgodności”;
4) poinformowanie pacjenta o ryzyku i korzyściach wynikających z przetoczenia – dotyczy wyłącznie lekarza;
5) identyfikacja biorcy, na podstawie danych, o których mowa w § 11 ust. 1 pkt 1, i kontrola dokumentacji medycznej przed przetoczeniem;
6) przetoczenie;
7) obserwacja pacjenta w trakcie przetoczenia i po przetoczeniu oraz podjęcie odpowiednich czynności, jeżeli wystąpi powikłanie.
2. Informacje o zabiegu przetoczenia i powikłaniach poprzetoczeniowych odnotowuje się w historii choroby, książce transfuzyjnej, której wzór jest określony w załączniku nr 1 do rozporządzenia, karcie informacyjnej z leczenia szpitalnego pacjenta oraz w księdze raportów pielęgniarskich.
3. Wpisów w książce transfuzyjnej dokonuje pielęgniarka, przy czym każdy wpis musi być sprawdzony przez lekarza odpowiedzialnego za przetoczenie i potwierdzony jego podpisem i imienną pieczątką.
2. W przypadku gdy w podmiocie leczniczym nie zatrudnia się lekarza specjalisty w dziedzinie transfuzjologii klinicznej, obowiązki lekarza odpowiedzialnego za gospodarkę krwią powierza się lekarzowi specjaliście, w szczególności w jednej z następujących dziedzin medycyny: chirurgii ogólnej, położnictwa i ginekologii, anestezjologii i intensywnej terapii, chorób wewnętrznych, hematologii, pediatrii lub onkologii klinicznej.
3. Lekarz odpowiedzialny za gospodarkę krwią:
1) odbywa przeszkolenie w jednej z jednostek organizacyjnych publicznej służby krwi, o których mowa w art. 4 ust. 3 ustawy, nie rzadziej niż co 4 lata;
2) bierze udział w kursach i seminariach organizowanych przez jednostki organizacyjne publicznej służby krwi, o których mowa w art. 4 ust. 3 ustawy.
4. Do zadań lekarza odpowiedzialnego za gospodarkę krwią należy w szczególności:
1) nadzór nad leczeniem krwią w jednostkach lub komórkach organizacyjnych przedsiębiorstwa podmiotu leczniczego;
2) planowanie zaopatrzenia podmiotu leczniczego w krew i jej składniki;
3) kierowanie bankiem krwi, jeżeli nie powierzono tej czynności kierownikowi pracowni serologii lub immunologii transfuzjologicznej;
4) zapewnienie przestrzegania standardowych procedur operacyjnych sporządzonych, w porozumieniu z jednostką organizacyjną publicznej służby krwi, o której mowa w art. 4 ust. 3 pkt 2–4 ustawy, zwaną dalej „centrum”, przez bank krwi, pracownię serologii lub immunologii transfuzjologicznej lub inne jednostki lub komórki organizacyjne przedsiębiorstwa podmiotu leczniczego;
5) organizacja wewnętrznych szkoleń lekarzy i pielęgniarek (położnych) w dziedzinie leczenia krwią w podmiocie leczniczym;
6) niezwłoczne przekazywanie do centrum raportów o zdarzeniach, o których mowa w § 2 pkt 3;
7) sporządzanie i przekazywanie do centrum rocznych sprawozdań z działalności podmiotu leczniczego w zakresie krwiolecznictwa, nie później niż do dnia 30 stycznia każdego roku, za rok poprzedni.
2. Zmiany w wykonywaniu danej procedury wprowadza się, sporządzając nową SOP lub nową wersję SOP.
3. Czynności określone w rozdziałach 2–4 opisuje się w SOP.
4. Kierownik podmiotu leczniczego przechowuje oryginał SOP.
5. Kopie SOP sporządza się dla każdego stanowiska pracy związanego z leczeniem krwią.
6. Kopie SOP, o których mowa w ust. 5, przechowuje kierownik jednostki lub komórki organizacyjnej przedsiębiorstwa podmiotu leczniczego.
7. SOP sporządza się według wzoru określonego w załączniku nr 2 do rozporządzenia.
8. SOP zatwierdza kierownik podmiotu leczniczego po akceptacji dyrektora centrum lub osoby przez niego upoważnionej.
2. Wpis dokonany w dokumentacji, o której mowa w ust. 1, nie może być z niej usunięty.
3. Poprawek lub zmian w dokumentacji należy dokonywać w taki sposób, aby możliwe było jednoznaczne rozróżnienie danych pierwotnych od danych, które uległy poprawieniu lub zmianie.
4. Wprowadzoną poprawkę lub zmianę należy każdorazowo opatrzyć podpisem osoby, która tę poprawkę lub zmianę wprowadziła, oraz datą jej dokonania.
1) rozwiązywania problemów dotyczących leczenia krwią i jej składnikami;
2) rozwiązywania problemów związanych z gospodarką krwią;
3) sprawowania nadzoru nad leczeniem krwią i jej składnikami.
2. W skład komitetu wchodzą:
1) ordynatorzy lub inne osoby kierujące jednostkami lub komórkami organizacyjnymi przedsiębiorstwa podmiotu leczniczego, w których często przetacza się krew, lub ich zastępcy;
2) lekarz odpowiedzialny za gospodarkę krwią;
3) anestezjolog;
4) kierownik pracowni serologii lub immunologii transfuzjologicznej;
5) pielęgniarka (położna) dokonująca przetoczeń;
6) kierownik banku krwi.
3. Do jednostek lub komórek organizacyjnych przedsiębiorstwa podmiotu leczniczego, o których mowa w ust. 2 pkt 1, zalicza się w szczególności oddziały: chirurgiczny, położniczy i ginekologiczny, ortopedyczny, kardiochirurgiczny, hematologiczny, pediatryczny, neonatologiczny, chorób wewnętrznych oraz intensywnej opieki medycznej.
4. Komitet współpracuje z jednostkami organizacyjnymi publicznej służby krwi, w szczególności z właściwym centrum.
5. Do zadań komitetu należy w szczególności:
1) dokonywanie okresowej oceny wskazań do przetoczenia, nie rzadziej niż raz na 6 miesięcy;
2) analiza zużycia krwi i jej składników oraz produktów krwiopochodnych w celu ograniczenia niepotrzebnych przetoczeń i nadmiernych zniszczeń tej krwi, jej składników oraz produktów;
3) nadzór nad działaniami związanymi z leczeniem krwią oraz nadzór nad związaną z tym dokumentacją;
4) ocena stosowanej metodyki przetoczeń określonej w rozporządzeniu;
5) analiza każdego powikłania poprzetoczeniowego wraz z oceną postępowania;
6) analiza raportów o zdarzeniach, o których mowa w § 2 pkt 3;
7) opracowanie programu kształcenia lekarzy i pielęgniarek (położnych) w dziedzinie leczenia krwią i nadzór nad jego realizacją;
8) udział w planowaniu zaopatrzenia w krew i jej składniki oraz w rocznej sprawozdawczości dotyczącej ich zużycia.
6. Raporty i okresowe sprawozdania z działalności komitetu są przekazywane kierownikowi podmiotu leczniczego i dyrektorowi właściwego centrum nie rzadziej niż raz na rok, najpóźniej do dnia 30 stycznia każdego roku.
Rozdział 2
Organizacja leczenia krwią w jednostkach lub komórkach organizacyjnych przedsiębiorstwa podmiotu leczniczego
§ 9. [Czynności przed przetoczeniem krwi]
2. Za wiarygodny można uznać wyłącznie wynik badania grupy krwi wpisany w karcie identyfikacyjnej grupy krwi, której wzór jest określony w załączniku nr 3 do rozporządzenia, lub wynik z pracowni serologii lub immunologii transfuzjologicznej.
3. Jeżeli badanie grupy krwi nie zostało przeprowadzone lub istnieją wątpliwości co do wiarygodności wyniku badania grupy krwi, przed wykonaniem przetoczenia należy pobrać próbkę krwi od pacjenta w celu ponownego przeprowadzenia badania.
4. Próbkę krwi pobiera się na podstawie wypełnionego i podpisanego przez lekarza zlecenia na badanie grupy krwi, którego wzór jest określony w załączniku nr 4 do rozporządzenia.
5. Na podstawie wyniku badania grupy krwi lekarz wypełnia zamówienie indywidualne na krew i jej składniki, którego wzór jest określony w załączniku nr 5 do rozporządzenia, który przekazuje się do banku krwi.
6. Jeżeli w podmiocie leczniczym nie ma banku krwi, zamówienie indywidualne na krew i jej składniki przekazuje się bezpośrednio do centrum.
7. W przypadku planowanego przetoczenia koncentratu krwinek czerwonych, zwanego dalej „KKCz”, krwi pełnej konserwowanej, zwanej dalej „KPK”, lub koncentratu granulocytarnego, zwanego dalej „KG”, do banku krwi przekazuje się zamówienie indywidualne na krew i jej składniki oraz zlecenie wykonania próby zgodności, którego wzór jest określony w załączniku nr 6 do rozporządzenia, wraz z odrębnie w tym celu pobraną próbką krwi od pacjenta.
8. Pracownik banku krwi sprawdza zgodność grupy krwi i numeru donacji na segmencie drenu z grupą krwi i numerem na etykiecie pojemnika i przekazuje segmenty drenów wraz ze zleceniem i próbką krwi, o których mowa w ust. 7, do pracowni serologii lub immunologii transfuzjologicznej.
9. W przypadkach bezpośredniego zagrożenia życia lekarz może podjąć decyzję o przetoczeniu KKCz albo KPK zgodnych w układzie ABO i RhD, przed wykonaniem próby zgodności. Dopuszcza się również przetoczenie KKCz lub KPK grupy O RhD minus.
10. W przypadku, o którym mowa w ust. 9, do banku krwi przekazuje się zlecenie na krew do pilnej transfuzji, którego wzór jest określony w załączniku nr 7 do rozporządzenia. Następnie lekarz wypełnia zlecenie na badanie grupy krwi ABO i RhD, jeżeli brak wyniku, oraz zlecenie wykonania próby zgodności. Do dalszego postępowania stosuje się przepisy § 36 ust. 8–11.
2. Bezpośrednio po pobraniu krwi, na etykiecie probówki, w obecności pacjenta, wpisuje się, na podstawie danych uzyskanych od pacjenta, a jeżeli jest to niemożliwe – na podstawie danych z historii choroby, w tym w szczególności karty gorączkowej, lub stosowanego w podmiocie leczniczym znaku identyfikacyjnego:
1) nazwisko i imię pacjenta (drukowanymi literami);
2) datę urodzenia pacjenta lub numer PESEL, jeżeli posiada;
3) datę i godzinę pobrania próbki krwi.
3. W przypadku braku możliwości uzyskania danych pacjenta, o których mowa w ust. 2, na etykiecie i na zleceniu na badanie grupy krwi należy wpisać symbol „NN” oraz numer księgi głównej i numer księgi oddziałowej, jeżeli jest nadany, lub niepowtarzalny numer identyfikacyjny pacjenta.
4. Po pobraniu próbki krwi i opisaniu probówki osoba pobierająca sprawdza, czy dane pacjenta są zgodne z danymi na etykiecie probówki z próbką krwi pacjenta, zleceniu na badanie grupy krwi oraz zleceniu na wykonanie próby zgodności i składa na tych zleceniach czytelny podpis oraz wpisuje datę i godzinę pobrania.
5. Pobrana próbka krwi jest niezwłocznie dostarczana do pracowni serologii lub immunologii transfuzjologicznej wraz ze zleceniem na badanie grupy krwi i zleceniem na wykonanie próby zgodności.
6. Dopuszcza się pobieranie krwi na sól kwasu wersenianowego (EDTA) od:
1) noworodków;
2) niemowląt;
3) małych dzieci;
4) pacjentów z niedokrwistością autoimmunohemolityczną (NAIH);
5) pacjentów, u których badania grupy krwi wykonuje się metodami automatycznymi.
1) identyfikacji pacjenta poprzez porównanie jego imienia i nazwiska, daty urodzenia lub numeru PESEL oraz grupy krwi z danymi zawartymi w wyniku próby zgodności, którego wzór jest określony w załączniku nr 8 do rozporządzenia;
2) porównaniu wyników badania grupy krwi zawartych w wyniku próby zgodności z grupą krwi na etykiecie pojemnika;
3) porównaniu numeru donacji krwi lub jej składnika z numerem zawartym w wyniku próby zgodności;
4) sprawdzeniu, czy jednostka krwi lub jej składnika została przygotowana zgodnie ze specjalnymi zaleceniami zawartymi w zamówieniu na krew i jej składniki;
5) sprawdzeniu daty ważności krwi lub jej składnika.
2. Lekarz i uprawniona do tego pielęgniarka (położna), którzy dokonali oceny zgodności krwi lub jej składnika z biorcą, składają swój podpis na wyniku próby zgodności.
3. W przypadku przetaczania składnika krwi niewymagającego przed podaniem wykonania próby zgodności, czyli osocza, koncentratu krwinek płytkowych, zwanego dalej „KKP”, oraz krioprecypitatu, lekarz umieszcza w historii choroby adnotację dotyczącą dokonanej oceny zgodności ze złożonym zamówieniem na krew i jej składniki.
4. Godzinę rozpoczęcia przetoczenia zawartości każdego pojemnika należy wpisać w książce transfuzyjnej, w wyniku próby zgodności oraz w protokole znieczulenia ogólnego, jeżeli obowiązuje, a na oddziale anestezjologii i intensywnej terapii – w karcie obserwacji.
5. W razie rozbieżności wykrytych podczas kontroli zgodności krwi lub jej składnika z danymi biorcy nie wolno przetaczać tej jednostki krwi lub tego składnika.
6. W przypadku, o którym mowa w ust. 5, krew lub jej składnik należy zwrócić do banku krwi wraz z protokołem zawierającym informację o przyczynie zwrotu i wynikiem próby zgodności. Krew ta nie może być ponownie dopuszczona do użytku.
7. W przypadku, o którym mowa w ust. 5, należy sporządzić raport o zdarzeniu, o którym mowa w § 2 pkt 3.
8. Wynik próby zgodności musi być dostępny podczas przetoczenia.
2. Pojedyncze jednostki krwi należy sukcesywnie pobierać z banku krwi.
3. W wyjątkowych przypadkach, jeżeli przewiduje się, że czas do rozpoczęcia przetoczenia będzie dłuższy niż 30 minut, krew należy przechowywać w zwalidowanej chłodziarce, przeznaczonej wyłącznie do tego celu, w temperaturze od 2°C do 6°C, przy czym temperaturę w chłodziarce należy sprawdzać i zapisywać co najmniej 3 razy w ciągu doby (co 8 godzin).
4. Przetoczenie KKP, rozmrożonego osocza oraz rozmrożonego krioprecypitatu należy rozpocząć niezwłocznie po ich otrzymaniu.
5. Planowane przetoczenia powinny odbywać się w okresie pełnej obsady lekarzy i pielęgniarek (położnych) jednostki lub komórki organizacyjnej przedsiębiorstwa podmiotu leczniczego.
6. Składniki krwi przetacza się za pomocą jednorazowych sterylnych zestawów.
7. Nie można przetaczać KKP i płynów infuzyjnych przez zestaw uprzednio użyty do przetaczania KPK lub KKCz.
8. Do przetoczeń niemowlętom służą specjalne zestawy. Jeżeli składnik krwi jest podawany strzykawką, należy zastosować specjalny filtr.
9. Używane pompy muszą mieć atest i wskazówki producenta, jak należy je stosować.
10. Ogrzewanie krwi można przeprowadzać wyłącznie w specjalistycznym urządzeniu zaopatrzonym w termometr i system alarmowy. Zaleca się ogrzewanie krwi, do temperatury nie wyższej niż 37°C, w przypadku:
1) dorosłych – jeżeli szybkość przetoczenia przekracza 50 ml/min;
2) dzieci – jeżeli szybkość przetoczenia przekracza 15 ml/min;
3) noworodków – w przetoczeniu wymiennym;
4) biorców z klinicznie znaczącymi przeciwciałami typu zimnego.
11. Nie można dodawać produktów leczniczych do przetaczanej krwi.
12. Nie można przetaczać jednej jednostki krwi pełnej lub KKCz dłużej niż 4 godziny, a jednej jednostki KKP, osocza lub krioprecypitatu – dłużej niż 30 minut.
13. Nie można po odłączeniu ponownie podłączać pacjentowi tego samego zestawu lub tego samego pojemnika ze składnikiem krwi.
14. Przez jeden zestaw można przetaczać, podczas jednego zabiegu, jedną jednostkę krwi lub jej składnika. Zestaw należy zmienić również w przypadku, gdy po zakończonym przetoczeniu podaje się płyny infuzyjne.
15. Składnik krwi niezużyty w całości nie może być przetoczony innemu pacjentowi.
16. Po przetoczeniu pojemniki z pozostałością po przetoczeniu wraz z zestawami do przetoczenia opisane nazwiskiem i imieniem pacjenta oraz datą i godziną przetoczenia należy przechowywać w temperaturze od 2°C do 6°C przez 72 godziny w specjalnie do tego celu przeznaczonej chłodziarce, a następnie zutylizować w sposób uniemożliwiający pozyskanie danych osobowych pacjenta przez osoby nieuprawnione.
17. Pojemniki, o których mowa w ust. 16, muszą być odpowiednio zabezpieczone przed rozlaniem i wtórnym zakażeniem, a chłodziarki przeznaczone do ich przechowywania muszą podlegać wstępnej i okresowej walidacji. Pomiar temperatury w chłodziarce powinien być przeprowadzany co najmniej 3 razy w ciągu doby (co 8 godzin) i dokumentowany.
2. Przed przetoczeniem, po 15 minutach od rozpoczęcia przetoczenia każdej jednostki krwi lub jej składnika oraz po jego zakończeniu należy zmierzyć i zarejestrować ciepłotę ciała, tętno i ciśnienie tętnicze krwi pacjenta.
3. Lekarz odpowiedzialny za przetoczenie lub wyznaczona przez niego pielęgniarka (położna) oraz lekarz przejmujący opiekę nad pacjentem są obowiązani do obserwacji pacjenta podczas przetoczenia i przez 12 godzin po jego zakończeniu.
4. Pacjent, na własną prośbę, może być wypisany z podmiotu leczniczego przed upływem 12 godzin.
5. Pacjenta należy pouczyć o konieczności niezwłocznego zgłoszenia każdego niepokojącego objawu, a w szczególności dreszczy, wysypki, zaczerwienienia skóry, duszności, bólu kończyn lub okolicy lędźwiowej.
6. W przypadku wystąpienia objawów lub zmian, mogących świadczyć o powikłaniu poprzetoczeniowym, należy wdrożyć odpowiednie postępowanie w zależności od stanu pacjenta, w trakcie lub po zakończonym przetoczeniu, zgodnie z przepisami § 14 ust. 3–5.
7. Należy zwrócić szczególną uwagę na pacjentów, którzy są nieprzytomni. Pogorszenie stanu ogólnego pacjenta, w szczególności w ciągu 15–20 minut od rozpoczęcia przetaczania każdej jednostki składnika krwi, może być objawem powikłania poprzetoczeniowego, o którym mogą świadczyć: spadek ciśnienia tętniczego, nieuzasadnione krwawienie, które może być następstwem rozsianego wykrzepiania wewnątrznaczyniowego, hemoglobinuria lub oliguria, które mogą być pierwszym objawem ostrej hemolitycznej reakcji poprzetoczeniowej.
1) reakcje hemolityczne;
2) zakażenia bakteryjne;
3) odczyn anafilaktyczny;
4) ostre poprzetoczeniowe uszkodzenie płuc (ang. Transfusion Related Acute Lung Injury – TRALI);
5) duszność poprzetoczeniową;
6) niehemolityczne reakcje gorączkowe;
7) wysypkę.
2. Do opóźnionych powikłań poprzetoczeniowych zalicza się w szczególności:
1) reakcje hemolityczne;
2) poprzetoczeniową skazę małopłytkową;
3) poprzetoczeniową chorobę przeszczep przeciw biorcy (ang. Transfusion Associated Graft versus Host Disease – TA-GvHD);
4) przeniesienie biologicznych czynników chorobotwórczych.
3. W przypadku wystąpienia u pacjenta objawów nasuwających podejrzenie wczesnego powikłania poprzetoczeniowego składnika krwi należy:
1) niezwłocznie przerwać przetoczenie i powiadomić lekarza;
2) odłączyć pojemnik ze składnikiem krwi wraz z zestawem do przetoczenia, utrzymując jednocześnie wkłucie do żyły, i powoli przetaczać choremu – przez nowy sterylny zestaw – 0,9% roztwór chlorku sodowego (NaCl) do czasu wdrożenia odpowiedniego leczenia;
3) zmierzyć pacjentowi ciepłotę ciała, tętno i ciśnienie tętnicze krwi;
4) dalsze postępowanie uzależniać od nasilenia i rodzaju objawów.
4. W przypadku gdy potwierdzi się podejrzenie, że objawy wskazują na ostre powikłanie poprzetoczeniowe, należy niezwłocznie:
1) sprawdzić dane na wszystkich pojemnikach z przetaczanymi składnikami, wyniki próby zgodności i wynik grupy krwi pacjenta oraz dane identyfikujące pacjenta, o których mowa w § 11 ust. 1 pkt 1;
2) pobrać próbki krwi od pacjenta z innego miejsca wkłucia niż miejsce, w którym dokonywano przetoczenia, w celu wykonania badań:
a) immunohematologicznych w zakresie ustalonym z pracownią badań konsultacyjnych centrum, a w przypadku podejrzenia TRALI – w zakresie ustalonym przez jednostkę organizacyjną publicznej służby krwi, o której mowa w art. 4 ust. 3 pkt 1 ustawy,
b) bakteriologicznych (na posiew); objętość próbki krwi i rodzaj pojemnika z podłożem bakteriologicznym określa pracownia bakteriologiczna wskazana przez centrum;
3) powiadomić pracownię serologii lub immunologii transfuzjologicznej, która wykonywała badania przed przetoczeniem; pracownia kontroluje dokumentację i ponownie wykonuje badania grupy krwi biorcy i krwi dobieranej do przetoczenia, a wyniki badań przekazuje do lekarza odpowiedzialnego za przetoczenie;
4) powiadomić właściwe centrum, pod którego nadzór merytoryczny podlega terytorialnie dany podmiot leczniczy, w celu dalszego przeprowadzania badań oraz powiadomić centrum, z którego otrzymano składniki krwi, jeżeli jest to inne centrum niż właściwe;
5) przesłać do działu immunologii transfuzjologicznej właściwego centrum:
a) wszystkie próbki krwi pacjenta oraz krwi dobieranej do przetoczenia, znajdujące się w pracowni serologii lub immunologii transfuzjologicznej,
b) próbki krwi pacjenta pobrane do badań serologicznych po przetoczeniu,
c) w przypadku podejrzenia odczynu TRALI – próbki wraz ze zgłoszeniem powikłania poprzetoczeniowego, którego wzór jest określony w załączniku nr 9 do rozporządzenia; dział immunologii transfuzjologicznej właściwego centrum przesyła próbki do diagnostyki w jednostce organizacyjnej publicznej służby krwi, o której mowa w art. 4 ust. 3 pkt 1 ustawy;
6) przesłać do pracowni bakteriologicznej wskazanej przez właściwe centrum:
a) pobrane po przetoczeniu próbki krwi pacjenta,
b) wszystkie pojemniki z resztkami przetaczanej krwi; pracownia bakteriologiczna po pobraniu z pojemników próbek krwi do badań przesyła pojemniki z resztkami przetaczanej krwi do działu immunologii transfuzjologicznej właściwego centrum;
7) w przypadku wystąpienia duszności poprzetoczeniowej – przeprowadzić badania w celu diagnostyki TRALI, czyli badanie radiologiczne klatki piersiowej i oznaczenie BNP (mózgowy peptyd natriuretyczny) w surowicy;
8) przesłać do właściwego centrum zgłoszenie powikłania poprzetoczeniowego wypełnione przez lekarza odpowiedzialnego za przetoczenie.
5. W przypadku gdy ciężkie objawy powikłań wystąpią po zakończonym przetoczeniu, należy powiadomić lekarza odpowiedzialnego za przetoczenie i postępować zgodnie z przepisami ust. 4 pkt 4–8.
6. Centrum rejestruje wszystkie powikłania poprzetoczeniowe na podstawie dokumentacji przekazanej przez podmiot leczniczy.
7. W przypadkach ciężkich powikłań dyrektor centrum lub upoważniony przez niego lekarz przeprowadza kontrolę postępowania przed przetoczeniem i podczas jego przeprowadzania oraz udziela wskazówek dotyczących postępowania po wystąpieniu powikłania.
8. Centrum właściwe dla podmiotu leczniczego, w którym wystąpiło powikłanie poprzetoczeniowe, uwzględnia go w danych statystycznych.
Rozdział 3
Organizacja banku krwi w przedsiębiorstwie podmiotu leczniczego
§ 15. [Bank krwi]
2. Bank krwi w podmiocie leczniczym należy zlokalizować w odrębnym pomieszczeniu albo na terenie pracowni serologii lub immunologii transfuzjologicznej lub medycznego laboratorium diagnostycznego.
3. Kierownikiem banku krwi jest lekarz odpowiedzialny za gospodarkę krwią lub kierownik pracowni serologii lub immunologii transfuzjologicznej.
2. Do zadań banku krwi należy w szczególności:
1) składanie zamówień na krew i jej składniki we właściwym centrum, zgodnie z zamówieniami jednostek lub komórek organizacyjnych przedsiębiorstwa podmiotu leczniczego;
2) odbiór krwi i jej składników;
3) przechowywanie krwi i jej składników do czasu ich wydania do jednostki lub komórki organizacyjnej przedsiębiorstwa podmiotu leczniczego;
4) wydawanie krwi i jej składników do jednostek lub komórek organizacyjnych przedsiębiorstwa podmiotu leczniczego;
5) prowadzenie dokumentacji:
a) przychodów i rozchodów krwi i jej składników,
b) zawierającej dane pozwalające na identyfikację dawcy i biorcy krwi lub jej składników: imię i nazwisko, datę urodzenia lub numer PESEL oraz grupę krwi;
6) prowadzenie sprawozdawczości zużycia krwi i jej składników;
7) przekazywanie sprawozdań, o których mowa w pkt 6, do właściwego centrum.
3. Kierownik banku krwi, w porozumieniu z dyrektorem centrum, sporządza SOP.
2. Nadzór nad czynnościami osób, o których mowa w ust. 1, sprawuje kierownik banku krwi.
2. Książka przychodów i rozchodów zawiera w szczególności następujące informacje:
1) datę i godzinę przychodu;
2) nazwę, numer donacji, grupę krwi, ilość składnika krwi, datę pobrania oraz datę ważności;
3) podpis osoby przyjmującej;
4) datę i godzinę rozchodu;
5) nazwę jednostki lub komórki organizacyjnej przedsiębiorstwa podmiotu leczniczego, do którego przekazano składnik krwi;
6) imię, nazwisko, datę urodzenia lub numer PESEL biorcy; w przypadku braku danych pacjenta, symbol „NN” wraz z numerem księgi głównej i numerem księgi oddziałowej, jeżeli jest nadany, lub niepowtarzalny numer identyfikacyjny;
7) podpis osoby wydającej składnik krwi.
3. Wyniki kontroli temperatury w chłodziarkach, zamrażarkach i w innym sprzęcie do termostatowania, przeznaczonych do przechowywania krwi i jej składników, dokumentuje się poprzez sporządzenie protokołu kontroli temperatury przechowywania krwi i jej składników.
4. Protokoły kontroli temperatury przechowywania krwi i jej składników oraz protokoły kontroli temperatury transportu krwi i jej składników należy przechowywać przez okres co najmniej 5 lat od dnia dokonania pomiarów.
2. Składając wstępne zamówienie telefoniczne, należy uzgodnić termin i sposób dostarczenia krwi lub jej składników do banku krwi albo ich odbioru z centrum.
3. Uzupełniając zapas krwi i jej składników, bank krwi jest obowiązany sporządzić pisemne zamówienie zbiorcze na krew i jej składniki, którego wzór jest określony w załączniku nr 10 do rozporządzenia, i uzyskać jego akceptację przez kierownika podmiotu leczniczego lub osobę przez niego upoważnioną i głównego księgowego. Akceptacja taka może mieć formę stałej akceptacji rocznej.
4. Zamówienia indywidualne oraz zamówienia zbiorcze na krew i jej składniki należy dostarczyć do centrum przed wydaniem odbiorcy krwi lub jej składników.
5. W przypadkach nagłych krew i jej składniki można wydawać na podstawie zamówienia telefonicznego, zamówienia przesłanego faksem lub zamówienia przesłanego pocztą elektroniczną, z zachowaniem bezpieczeństwa danych. Zamówienie to musi być niezwłocznie uzupełnione formalnym złożeniem zamówienia na krew i jej składniki, najpóźniej przy odbiorze krwi lub jej składnika.
6. Zamówienia indywidualne oraz zamówienia zbiorcze na krew i jej składniki sporządza się w dwóch egzemplarzach. Oryginał zamówienia przesyła się do centrum, z którego otrzymuje się krew lub jej składniki, a kopię przechowuje się w banku krwi zgodnie z przepisami § 18 ust. 1.
2. Temperaturę mierzy się za pomocą termometru lub czujnika automatycznego.
3. Centrum, które wydało krew i jej składniki, oraz ich odbiorca sporządzają protokół kontroli transportu, który zawiera w szczególności następujące informacje:
1) nazwę i adres centrum wydającego krew i jej składniki;
2) nazwę i numer składnika;
3) dzień i godzinę wydania;
4) temperaturę odczytaną po 5 minutach od chwili umieszczenia krwi lub jej składnika w pojemniku transportowym;
5) opis chłodniczego urządzenia transportowego, z podaniem ilości i rodzaju dodatkowego materiału chłodzącego oraz numeru termometru – jeżeli je stosowano;
6) datę, podpis oraz pieczątkę osoby wydającej krew lub jej składnik;
7) imię i nazwisko osoby transportującej krew;
8) nazwę i adres podmiotu leczniczego będącego odbiorcą;
9) dzień i godzinę dostarczenia składnika krwi;
10) temperaturę odczytaną w chwili dostarczenia krwi lub jej składnika;
11) datę, podpis oraz pieczątkę osoby dokonującej odbioru krwi lub jej składnika.
4. W przypadku stosowania automatycznych czujników temperatury, do protokołu kontroli temperatury transportu zamiast danych, o których mowa w ust. 3 pkt 4 i 10, dołącza się wydruki otrzymane z czujników.
5. Protokół kontroli transportu sporządza się w dwóch egzemplarzach. Oryginał zatrzymuje odbiorca, a kopię – dostawca.
6. Jeżeli krew lub jej składniki są przewożone transportem podmiotu leczniczego, bank krwi jest odpowiedzialny za sporządzenie protokołu kontroli temperatury transportu krwi i jej składników.
1) zgodności etykiet z zamówieniem na krew i jej składniki;
2) daty ważności;
3) szczelności pojemników;
4) wyglądu krwi lub jej składników.
2. Przyjęcie pojemników z krwią lub jej składnikami potwierdza się przez umieszczenie na kopii kwitu rozchodowego centrum daty, podpisu i pieczątki kierownika banku krwi lub osoby upoważnionej do odbioru.
2. Urządzenie do przechowywania krwi i jej składników powinno być wyposażone w co najmniej dwa niezależne mierniki temperatury poddawane okresowej kalibracji lub wzorcowaniu, zgodnie z zaleceniami producenta.
3. Temperaturę kontroluje się i dokumentuje co najmniej 3 razy w ciągu doby (co 8 godzin).
4. W miarę możliwości należy używać urządzeń do termostatowania, w tym sprzętu chłodniczego i inkubatorów do przechowywania KKP, wyposażonych w alarm dźwiękowy i wizualny.
5. Temperaturę urządzeń, o których mowa w ust. 4, kontroluje się w sposób ciągły przy pomocy zapisu graficznego lub przy pomocy automatycznego wydruku okresowego. W przypadku gdy jest to niemożliwe, kontrolę prowadzi się na podstawie wskazań umieszczonych wewnątrz czujników temperatury i systematycznie dokumentuje, co najmniej 3 razy w ciągu doby (co 8 godzin), potwierdzając podpisem osoby dokonującej pomiaru.
6. Jeżeli urządzenie do przechowywania krwi i jej składników jest wyposażone w alarm, personel obsługujący to urządzenie powinien być poinformowany o:
1) dopuszczalnym zakresie temperatur;
2) temperaturach, przy których uruchamia się alarm (temperatury progowe);
3) czasie, po którym włącza się alarm.
7. Dla urządzenia do przechowywania krwi i jej składników prowadzi się dokumentację temperatur. Dokumentacja ta zawiera:
1) numer identyfikacyjny urządzenia;
2) zakres dopuszczalnej temperatury, wynikający z przeprowadzonej walidacji urządzeń do termostatowania;
3) numer identyfikacyjny urządzenia pomiarowego, w szczególności sondy;
4) datę i godzinę odczytu;
5) wartość temperatury wskazywanej przez dwa mierniki;
6) podpis osoby kontrolującej temperaturę.
8. W przypadku stwierdzenia nieprawidłowej temperatury sporządza się protokół wyjaśniający przyczynę zaistniałej sytuacji oraz opisuje podjęte kroki zaradcze, zgodnie z procedurami awaryjnymi.
9. Urządzenia do przechowywania krwi i jej składników podlegają wstępnej walidacji oraz okresowej, ponownej walidacji. Walidację oraz ponowną, okresową walidację przeprowadza bank krwi we własnym zakresie przy użyciu atestowanych mierników temperatury lub zleca przeprowadzenie tych czynności autoryzowanemu serwisowi producenta.
10. KPK oraz KKCz przechowuje się w temperaturze od 2°C do 6°C, w przeznaczonych wyłącznie do tego celu chłodniach lub chłodziarkach, przy czym:
1) składniki segreguje się według grup krwi ABO i RhD;
2) krew i składniki każdej grupy krwi, w miarę możliwości, należy przechowywać w osobnym urządzeniu chłodniczym;
3) każdy pojemnik umieszcza się w pozycji pionowej, w taki sposób, aby zapewnić swobodną cyrkulację powietrza pomiędzy pojemnikami.
11. Miejsca przeznaczone do przechowywania krwi i jej składników według grup krwi ABO i RhD należy oznakować w sposób trwały.
12. Krew przeznaczoną do przetoczeń autologicznych przechowuje się w wydzielonym, wyraźnie oznakowanym miejscu.
13. Osocze i krioprecypitat przechowuje się w zamrażarkach lub mroźniach, w temperaturze od –18°C do –25°C nie dłużej niż przez trzy miesiące od dnia pobrania, a w temperaturze –25°C lub niższej, nie dłużej niż przez 36 miesięcy od dnia pobrania.
14. Składniki wymienione w ust. 13 segreguje się według grup ABO i rodzaju składnika i przechowuje oddzielnie, w miejscach wyraźnie oznakowanych zgodnie z grupą krwi ABO.
15. KKP przechowuje się w temperaturze od 20°C do 24°C przy stałym mieszaniu w mieszadle obrotowym lub horyzontalnym.
2. Osoba upoważniona do wydawania krwi i jej składników jest obowiązana każdorazowo do dokonania oceny makroskopowej wydawanych składników:
1) szczelności pojemnika;
2) zmiany zabarwienia zawartości.
3. Dokonując oceny makroskopowej KKCz i KPK, należy zwrócić szczególną uwagę na:
1) oznaki hemolizy;
2) obecność skrzepów;
3) kolor zawartości.
4. Dokonując oceny makroskopowej rozmrożonego osocza i krioprecypitatu, należy zwrócić szczególną uwagę na:
1) zmętnienie;
2) obecność skrzepów.
5. Dokonując oceny makroskopowej KKP, należy zwrócić szczególną uwagę na agregaty.
6. Przed wydaniem krwi lub jej składników do jednostek lub komórek organizacyjnych przedsiębiorstwa podmiotu leczniczego należy dokładnie sprawdzić zgodność danych na etykiecie z zamówieniem na krew i jej składniki, a w szczególności porównać numer składnika z numerem na wyniku próby zgodności, jeżeli obowiązuje jej wykonanie.
7. Krew oraz jej składniki powinny być wydawane do jednostek lub komórek organizacyjnych przedsiębiorstwa podmiotu leczniczego bezpośrednio przed planowanym przetoczeniem po wykonaniu próby zgodności, jeżeli obowiązuje jej wykonanie, i innych wymaganych badań immunohematologicznych.
8. Składniki krwi wymagające rozmrożenia przed przetoczeniem (osocze, krioprecypitat) należy wydawać do jednostek lub komórek organizacyjnych przedsiębiorstwa podmiotu leczniczego w stanie płynnym.
9. Osocze i krioprecypitat rozmraża się w temperaturze 37°C, przy stałej kontroli temperatury.
10. Osocze i krioprecypitat rozmraża się przy użyciu specjalistycznych urządzeń, do których należą suche podgrzewacze oraz łaźnie wodne; suche podgrzewacze elektronicznie regulują ilość dostarczanego ciepła, utrzymując stałą temperaturę rozmrażania, oraz automatycznie przerywają podgrzewanie po osiągnięciu zaprogramowanej temperatury; łaźnie wodne z termoregulatorem są wyposażone w mieszadło zapewniające równomierny rozkład temperatury wody.
11. Przed umieszczeniem w urządzeniu, o którym mowa w ust. 10, pojemnik ze składnikiem należy szczelnie zamknąć w torebce foliowej; w przypadku zastosowania łaźni wodnej pojemnik należy umieścić w uchwycie w pozycji pionowej, aby zabezpieczyć go przed możliwością zakażenia drobnoustrojami przenoszonymi przez wodę.
12. Do wykonania próby zgodności należy pobierać z banku krwi wyłącznie segmenty drenów. Przed odcięciem segmentów należy sprawdzić zgodność zapisu grupy krwi i numeru składnika na pojemniku z krwią i na pobranym segmencie.
1) zgonu pacjenta, dla którego zamawiano krew lub jej składniki;
2) przypadku rzadko występującego fenotypu krwinek czerwonych;
3) innego uzasadnionego przypadku – po wyrażeniu zgody przez dyrektora centrum.
2. Krew lub jej składnik można zwrócić, pod warunkiem że:
1) dana jednostka krwi lub jej składniki były przechowywane i transportowane we właściwy sposób przy zachowaniu odpowiedniej i prawidłowo kontrolowanej temperatury oraz przy użyciu zwalidowanego sprzętu chłodniczego;
2) w podmiocie leczniczym, z którego krew jest przyjmowana, centrum przeprowadziło wcześniej kontrolę potwierdzoną protokołem, stwierdzającym brak uchybień w stosunku do obowiązujących przepisów dotyczących przechowywania krwi i jej składników.
3. Zwrot krwi lub jej składnika może być przyjęty na podstawie prawidłowo wypełnionego:
1) protokołu niewykorzystania krwi lub jej składników, który powinien zawierać:
a) nazwę i adres banku krwi podmiotu leczniczego dokonującego zwrotu,
b) nazwę, numer donacji, ilość i grupę krwi zwracanego składnika,
c) przyczynę niewykorzystania składnika,
d) datę i godzinę pobrania składnika krwi z centrum,
e) datę i godzinę dokonania zwrotu do centrum,
f) imię, nazwisko, pieczątkę i podpis kierownika banku krwi lub osoby przez niego upoważnionej oraz osoby dokonującej zwrotu;
2) protokołu kontroli temperatury przechowywania krwi i jej składników, który powinien zawierać co najmniej następujące dane:
a) nazwę, numer i grupę krwi składnika,
b) nazwę banku krwi, w którym przechowywano składnik krwi,
c) warunki przechowywania:
– temperaturę przechowywania,
– nazwę i numer chłodziarki lub zamrażarki, lub inkubatora (jeżeli dotyczy),
– czas przechowywania w chłodziarce, zamrażarce lub inkubatorze,
– kopie protokołów kontroli temperatury przeprowadzonej w okresie przechowywania składnika,
– datę i numer protokołu z ostatniej walidacji urządzenia, które wykorzystano do przechowywania składnika, lub specjalistyczny wskaźnik na pojemniku, potwierdzający prawidłowe warunki przechowywania i transportu,
d) datę, podpis i pieczątkę osoby sporządzającej protokół;
3) protokołu kontroli temperatury transportu krwi i jej składników, który sporządza się w przypadku, gdy krew i jej składniki nie były przewożone środkami transportu kontrolowanymi przez centrum; protokół ten zawiera co najmniej następujące dane, dotyczące, w razie potrzeby, warunków transportu w obie strony:
a) nazwę i adres banku krwi zamawiającego albo zwracającego składnik,
b) nazwę, numer i grupę krwi składnika,
c) czas trwania transportu,
d) warunki transportu:
– temperaturę,
– dokładny opis urządzenia zapewniającego właściwą temperaturę transportu,
– kopię protokołu kontroli transportu krwi lub jej składnika z centrum, które wydało krew lub składnik krwi, do odbiorcy,
– datę i numer protokołu z ostatniej walidacji urządzenia, którego użyto do transportu składnika krwi,
e) datę, podpis i pieczątkę osoby sporządzającej protokół.
Rozdział 4
Organizacja pracowni serologii lub immunologii transfuzjologicznej w przedsiębiorstwie podmiotu leczniczego
§ 26. [Stosowanie przepisów laboratoriów i pracowni wykonujących badania immunohematologiczne]
2. W strukturze przedsiębiorstwa podmiotu leczniczego pracownia serologii lub immunologii transfuzjologicznej, o której mowa w ust. 1, działa jako:
1) samodzielna jednostka albo komórka organizacyjna;
2) wydzielona pracownia wchodząca w skład medycznego laboratorium diagnostycznego;
3) samodzielna jednostka albo komórka organizacyjna połączona z bankiem krwi;
4) wydzielona pracownia wchodząca w skład medycznego laboratorium diagnostycznego połączona z bankiem krwi.
3. Podmiot leczniczy zapewnia pracowni serologii lub immunologii transfuzjologicznej:
1) odpowiednie pomieszczenie do wykonywania badań, w którym temperatura powinna wynosić 18–25°C;
2) odpowiedni sprzęt, aparaturę, odczynniki diagnostyczne, krwinki wzorcowe oraz formularze, których wzory są określone w załącznikach do rozporządzenia.
2. Kierownik pracowni serologii lub immunologii transfuzjologicznej musi posiadać zaświadczenie upoważniające do samodzielnego wykonywania i autoryzowania badań w zakresie immunologii transfuzjologicznej, wydane przez dyrektora jednej z jednostek organizacyjnych publicznej służby krwi, o których mowa w art. 4 ust. 3 ustawy, oraz co najmniej dwuletnią praktykę w wykonywaniu badań immunohematologicznych.
3. Organizacja pracy pracowni, za którą odpowiada jej kierownik, musi zapewnić gotowość do wykonywania badań immunohematologicznych przez całą dobę, gwarantując bezpieczeństwo biorców krwi, w tym noworodków zagrożonych chorobą hemolityczną (konflikt serologiczny).
2. Zaświadczenie upoważniające do samodzielnego wykonywania badań immunohematologicznych można wydać diagnoście laboratoryjnemu, technikowi analityki medycznej oraz lekarzowi, posiadającym co najmniej roczną praktykę w wykonywaniu tych badań, odbytą w pełnym wymiarze czasu pracy lub w niepełnym wymiarze czasu pracy odpowiadającym rocznemu pełnemu wymiarowi czasu pracy przez okres nie dłuższy niż trzy lata.
3. Personel zatrudniony w pracowni serologii lub immunologii transfuzjologicznej, co najmniej raz w roku, jest poddawany kontroli jakości wykonywanych badań, która jest prowadzona przez właściwe centrum. Wynik dokonanej oceny jest przekazywany kierownikowi pracowni serologii lub immunologii transfuzjologicznej podmiotu leczniczego. W przypadku gdy przekazana ocena jest negatywna, kierownik podmiotu leczniczego podejmuje, niezwłocznie, działania mające na celu zapewnienie wymaganej jakości wykonywanych badań.
4. W przypadku przerwy w wykonywaniu pracy w pracowni serologii lub immunologii transfuzjologicznej dłuższej niż 1 rok personel zatrudniony w pracowni serologii lub immunologii transfuzjologicznej musi odbyć dodatkowe szkolenie w jednostce organizacyjnej publicznej służby krwi, o której mowa w art. 4 ust. 3 ustawy.
5. Liczba osób zatrudnionych w pracowni serologii lub immunologii transfuzjologicznej jest uzależniona od zakresu i liczby wykonywanych badań, ale nie może być mniejsza niż dwie osoby.
6. Personel wykonujący badania immunohematologiczne w pracowni serologii lub immunologii transfuzjologicznej nie może być przemieszczany na stanowiska pracy w innych pracowniach.
7. Do wykonywania badań poza podstawowymi godzinami funkcjonowania podmiotu leczniczego, określonymi w regulaminie organizacyjnym podmiotu leczniczego, dopuszcza się jedną osobę – diagnostę laboratoryjnego, lekarza lub technika analityki medycznej, która ma aktualne zaświadczenie z centrum upoważniające do wykonywania badań.
8. Przebieg pracy w pracowni serologii lub immunologii transfuzjologicznej należy odnotowywać w książce raportów. W szczególności należy uwzględnić: godziny przyjęcia próbek krwi oraz wydania wyników badań, napotykane trudności z wydawaniem krwi do pilnej transfuzji. Raporty codziennie powinny być przeglądane, analizowane i podpisywane przez kierownika pracowni serologii lub immunologii transfuzjologicznej lub przez inną upoważnioną przez niego osobę.
9. Wszystkie wyniki badań wydawane z pracowni serologii lub immunologii transfuzjologicznej muszą być autoryzowane przez diagnostę laboratoryjnego lub lekarza posiadających zaświadczenie upoważniające do wykonywania badań i autoryzacji wyników.
1) cieplarkę, blok grzewczy z regulacją temperatury w granicach 18–72°C;
2) wirówki laboratoryjne z wirnikiem horyzontalnym o maksymalnym przyspieszeniu 3850 x g i z regulacją czasu wirowania;
3) chłodziarkę laboratoryjną z możliwością ustawienia temperatury 2–8°C, z zamrażalnikiem do przechowywania próbek surowic i osocza, głównie od pacjentów z alloprzeciwciałami.
2. Zalecane wyposażenie pracowni serologii lub immunologii transfuzjologicznej w aparaturę obejmuje:
1) automaty do badań serologicznych grup krwi i prób zgodności z oprogramowaniem dla pracowni serologii lub immunologii transfuzjologicznej i banku krwi;
2) aparaturę do testów mikrokolumnowych lub innych – zalecana w pracowni, która nie wykonuje badań metodą automatyczną;
3) oprogramowanie dla pracowni serologii lub immunologii transfuzjologicznej i banku krwi;
4) wirówkę do automatycznego przemywania krwinek w probówkowych testach antyglobulinowych; wirówka jest konieczna, jeżeli nie są stosowane automaty do badań grup krwi lub testy mikrokolumnowe lub inne;
5) komputer stanowiskowy.
3. Chłodziarkę, cieplarkę i blok grzewczy należy wyposażyć w dwa niezależne mierniki temperatury.
4. Temperaturę aparatury, o której mowa w ust. 3, kontroluje się w sposób ciągły przy pomocy zapisu graficznego lub automatycznego wydruku okresowego, a gdy jest to niemożliwe, kontrolę prowadzi się na podstawie wskazań umieszczonych wewnątrz czujników temperatury i systematycznie dokumentuje, co najmniej 3 razy w ciągu doby (co 8 godzin), potwierdzając podpisem osoby dokonującej pomiaru.
1) opiniuje obsadę stanowiska kierownika pracowni oraz liczbę niezbędnych do zatrudnienia osób w zależności od zakresu i liczby wykonywanych badań;
2) szkoli pracowników pracowni i dopuszcza ich do prowadzenia badań;
3) przeprowadza weryfikację pracowników w przypadku co najmniej rocznej przerwy w wykonywaniu badań;
4) przeprowadza, co najmniej raz na dwa lata, okresowe kontrole organizacji pracy, stosowanych metod i procedur oraz wyposażenia i warunków pracy;
5) wydaje zalecenia oraz nadzoruje ich wykonanie w przypadku stwierdzenia nieprawidłowości;
6) zaleca wprowadzenie uzasadnionych zmian w funkcjonowaniu pracowni oraz modyfikację technik serologicznych, dokumentacji i formularzy.
2. Protokół z kontroli przeprowadzonej przez centrum przekazuje się kierownikowi podmiotu leczniczego i kierownikowi pracowni serologii lub immunologii transfuzjologicznej.
1) imię i nazwisko pacjenta;
2) datę urodzenia pacjenta lub numer PESEL;
3) datę i godzinę pobrania próbki krwi.
2. Pracownia serologii lub immunologii transfuzjologicznej przyjmuje do badań tylko próbki krwi wraz z dołączonym zleceniem na badanie grupy krwi lub zleceniem na wykonanie grupy zgodności.
3. W przypadku braku danych pacjenta do badania można przyjąć próbki krwi i zlecenia z wpisanym symbolem „NN”, numerem księgi głównej, numerem księgi oddziałowej lub niepowtarzalnym numerem identyfikacyjnym.
4. Po otrzymaniu próbek krwi należy sprawdzić zgodność danych na etykietach probówek i zleceniach. Jeżeli dane na probówce są niezgodne z danymi na zleceniu, należy wyjaśnić przyczynę rozbieżności i w razie potrzeby zażądać ponownego pobrania krwi od pacjenta.
5. Próbki krwi należy zarejestrować w książce badań grup krwi, której wzór jest określony w załączniku nr 11 do rozporządzenia, lub w książce prób zgodności, której wzór jest określony w załączniku nr 12 do rozporządzenia, nadając im kolejne numery.
6. W przypadku biorców leczonych krwią w przeszłości i w przypadku kobiet z ciążami w wywiadzie, należy zapoznać się z całą dostępną dokumentacją dotyczącą poszukiwania i identyfikacji przeciwciał, która może zawierać także dane o powikłaniach poprzetoczeniowych.
7. Po wykonaniu wszystkich badań i wydaniu wyników, próbki krwi pacjentów i dawców przechowuje się w temperaturze od 2°C do 8°C przez 5 dni.
1) określanie grupy krwi ABO i RhD;
2) przeglądowe badanie na obecność przeciwciał odpornościowych do antygenów krwinek czerwonych w pośrednim teście antyglobulinowym;
3) próba zgodności krwi;
4) badanie w kierunku konfliktu serologicznego między matką a płodem i w chorobie hemolitycznej płodu lub noworodka;
5) badanie kwalifikujące do podania immunoglobuliny anty-D w ramach profilaktyki konfliktu serologicznego RhD;
6) badanie w celu określania fenotypu krwinek czerwonych i identyfikacja przeciwciał;
7) badanie grupy krwi w celu trwałej ewidencji.
2. W przypadku trudności w określeniu grupy krwi ABO należy skonsultować się niezwłocznie z centrum. W sytuacjach nagłych, do czasu ich wyjaśnienia, można dobierać do przetoczenia KKCz grupy O.
3. U pacjentów, w tym u kobiet w ciąży, u których określa się grupy krwi, wykonuje się przeglądowe badanie w kierunku przeciwciał odpornościowych w pośrednim teście antyglobulinowym. Po uzyskaniu dodatniego wyniku w badaniu przeglądowym dokonuje się identyfikacji przeciwciał.
4. Jeżeli pracownia serologii lub immunologii transfuzjologicznej nie ma możliwości wykonania badań, o których mowa w ust. 3, przesyła próbki do centrum wraz ze zleceniem na konsultacyjne badania immunohematologiczne, którego wzór jest określony w załączniku nr 13 do rozporządzenia, i protokołem wykonanych badań.
5. Oryginał wyniku badania w centrum przekazuje się pracowni serologii lub immunologii transfuzjologicznej podmiotu leczniczego, a wynik wpisuje się w książce badań grup krwi.
6. Jedną kopię wyniku badania w centrum przekazuje się do jednostki lub komórki organizacyjnej przedsiębiorstwa podmiotu leczniczego w celu wydania pacjentowi, a drugą kopię wyniku przekazuje się lekarzowi prowadzącemu leczenie pacjenta w celu umieszczenia w historii choroby.
7. U biorców systematycznie leczonych krwią oraz u osób, którym przetaczano krew w okresie ostatnich 3 miesięcy, bezwzględnie należy przestrzegać czasu ważności próby zgodności, który – liczony od momentu pobrania próbki krwi od pacjenta – wynosi 48 godzin. Jeżeli krew nie została w tym czasie przetoczona, należy powtórzyć próbę zgodności ze świeżo pobraną próbką krwi pacjenta.
8. Próbę zgodności wykonuje się z próbki krwi biorcy pobranej wyłącznie w tym celu. Jako krew dawcy służy próbka zawarta w segmencie drenu połączonego z pojemnikiem z krwią. Przed odłączeniem segmentu drenu należy porównać jego numer donacji z numerem na etykiecie pojemnika.
9. Wyniki próby zgodności wpisuje się na formularzu wyniku próby zgodności.
10. Wzór zlecenia na wykonanie badań immunohematologicznych kwalifikujących do podania immunoglobuliny anty-D w ramach profilaktyki konfliktu serologicznego RhD jest określony w załączniku nr 14 do rozporządzenia. Wzór wyniku tych badań jest określony w załączniku nr 15 do rozporządzenia.
2. Wyniki badań grup krwi i prób zgodności wykonanych przy zastosowaniu technik manualnych odczytuje się i wpisuje z udziałem dwóch osób, przy czym jedna z nich musi posiadać zaświadczenie uprawniające do samodzielnego wykonywania badań immunohematologicznych.
3. Każde wpisane badanie zawiera ostateczny wynik i jest czytelnie podpisywane przez osobę wykonującą i autoryzującą wynik.
4. Wpisy z danej serii badań podpisuje osoba wykonująca badanie i diagnosta laboratoryjny autoryzujący wyniki.
5. Po wpisaniu wyniku badania grupy krwi w wyniku badania grupy krwi, którego wzór jest określony w załączniku nr 16 do rozporządzenia, kierownik pracowni serologii lub immunologii transfuzjologicznej lub upoważniona przez niego osoba sprawdza, czy dane z książki badań grup krwi zgadzają się z danymi na wyniku badania grupy krwi, i składa podpis w książce i na wyniku.
6. Jeżeli badania wykonuje się technikami automatycznymi, należy prowadzić dokumentację w postaci wydruków komputerowych zamiast książek, o których mowa w ust. 1.
2. Pacjentom, u których wykryto obecność autoprzeciwciał reagujących w temperaturze 37°C, należy dobierać krew zgodną fenotypowo w układzie Rh i antygenie K z układu Kell.
3. Dziewczętom i kobietom do okresu menopauzy wskazane jest dobieranie krwi K ujemnej (K–) w ramach profilaktyki konfliktu serologicznego. Jeżeli wykonano badania antygenu K i stwierdzono jego obecność na krwinkach, można przetaczać krew K dodatnią (K+).
4. Biorcom, u których wykryto przeciwciała odpornościowe w aktualnym badaniu lub w przeszłości, dobiera się krew niezawierającą odpowiadającego im antygenu oraz zgodną fenotypowo w układzie Rh i antygenie K z układu Kell.
5. Dopuszcza się przetaczanie KKCz grupy O pacjentom innej grupy krwi w przypadku:
1) braku krwi jednoimiennej w układzie ABO;
2) bardzo słabej ekspresji antygenu A lub B albo trudności w oznaczeniu grupy krwi ABO.
6. Dopuszcza się przetaczanie KKCz grupy A lub B biorcom grupy AB, gdy brak jest krwi jednoimiennej.
7. Biorcy RhD dodatniemu można przetaczać KKCz RhD dodatni lub RhD ujemny.
8. W przypadku bezpośredniego zagrożenia życia, na pisemne zlecenie na krew do pilnej transfuzji wydane przez lekarza prowadzącego leczenie lub lekarza odpowiedzialnego za gospodarkę krwią, można wydać zamówioną krew zgodną w układzie ABO i RhD przed wykonaniem próby zgodności.
9. Po wydaniu krwi, zgodnie z ust. 8, przystępuje się niezwłocznie do wykonania próby zgodności. Jeżeli wynik wskazuje na niezgodność, konieczne jest natychmiastowe powiadomienie o tym lekarza prowadzącego leczenie, w celu przerwania przetoczenia.
10. W wyjątkowo nagłym przypadku, gdy lekarz prowadzący leczenie zdecyduje o przetoczeniu przed wykonaniem badania grupy krwi u biorcy i próby zgodności, należy wydać KKCz grupy O, a dziewczętom i kobietom w okresie rozrodczym – KKCz grupy O RhD ujemny, K ujemny.
11. W przypadku, o którym mowa w ust. 10, należy natychmiast przystąpić do określenia u biorcy grupy krwi ABO i RhD, wykonania przeglądowego badania przeciwciał odpornościowych i próby zgodności. Do dalszych przetoczeń należy kwalifikować krew jednoimienną z biorcą w układzie ABO i RhD.
1) określić grupę krwi w układzie ABO i antygen D z układu Rh u matki i u dziecka;
2) wykonać badania w kierunku obecności alloprzeciwciał odpornościowych w surowicy matki;
3) wykonać bezpośredni test antyglobulinowy (BTA) u dziecka.
2. Noworodkom i niemowlętom do ukończenia 4 miesiąca życia grupy A lub B urodzonym przez matki innej grupy przetacza się krwinki grupy O. Nie dotyczy to dzieci urodzonych przez matki grupy AB.
3. Dziecku innej grupy krwi ABO niż matka można przetaczać krew jednoimienną z jego grupą krwi, pod warunkiem wykazania nieobecności odpornościowych przeciwciał anty-A lub anty-B klasy IgG lub po wykonaniu próby krzyżowej z surowicą dziecka.
4. W przypadku gdy matka jest grupy AB, dziecku należy przetaczać krwinki jednoimienne z jego grupą krwi.
5. Krew grupy jednoimiennej w układzie ABO z dzieckiem można przetaczać wyłącznie po wykazaniu nieobecności przeciwciał anty-A lub anty-B klasy IgG u matki lub u dziecka.
6. Jeżeli w surowicy matki nie wykrywa się odpornościowych alloprzeciwciał, a u noworodka lub niemowlęcia do ukończenia 4 miesiąca życia BTA jest ujemny, do przetoczenia wydaje się krew zgodną z krwinkami tego noworodka lub niemowlęcia w układzie ABO i w RhD, bez próby zgodności, pod warunkiem sprawdzenia we krwi przeznaczonej do przetoczenia grupy ABO i antygenu D.
7. Jeżeli u matki wykryto przeciwciała odpornościowe, noworodkowi lub niemowlęciu do ukończenia 4 miesiąca życia dobiera się krew bez antygenu, do którego są skierowane; przed każdym przetoczeniem należy wykonywać próbę zgodności z surowicą matki.
8. Jeżeli krew matki jest niedostępna, należy sprawdzić obecność odpornościowych przeciwciał w surowicy noworodka lub niemowlęcia do ukończenia 4 miesiąca życia oraz przed pierwszym przetoczeniem należy wykonać próbę zgodności.
9. Ujemny wynik badania przeglądowego na obecność przeciwciał i ujemny wynik w próbie zgodności upoważniają do odstąpienia od wykonywania tych badań przed następnymi przetoczeniami.
1) należy dobierać krwinki czerwone grupy O niezawierające antygenów, do których są skierowane przeciwciała wykryte u matki;
2) w celu uniknięcia uodpornienia ciężarnej antygenami zawartymi w krwi użytej do przetoczenia dopłodowego należy oznaczyć u niej antygeny z układu Kell (K i k), Kidd (Jka i Jkb), Duffy (Fya i Fyb), MNS (S i s) i dobierać do przetoczenia krwinki czerwone dawców zgodne z krwią matki;
3) jeżeli grupa krwi ABO płodu jest zgodna z grupą krwi matki oraz gdy matka ma grupę AB, a płód ma grupę A lub B, można dobierać krwinki jednoimienne w układzie ABO z dzieckiem.
1) w konflikcie RhD należy dobierać krwinki czerwone RhD ujemne, przy czym:
a) w przypadku gdy zachodzi zgodność serologiczna w układzie ABO między matką i dzieckiem, przetacza się krew pełną rekonstytuowaną, zwaną dalej „KPR”, składającą się z KKCz i osocza od dawców o zgodnej grupie krwi w układzie ABO z dzieckiem,
b) noworodkom o grupie krwi A lub B matek innej grupy krwi w układzie ABO niż dziecko przetacza się KPR, składającą się z KKCz od dawcy grupy O, zawieszonego w osoczu od dawcy grupy zgodnej z grupą krwi dziecka lub w osoczu grupy AB; nie dotyczy to dzieci urodzonych przez matki grupy AB;
2) przy konflikcie w układzie ABO należy dobrać KPR, składającą się z KKCz od dawcy grupy O i RhD, zgodnym z RhD dziecka, zawieszonego w osoczu od dawcy grupy krwi ABO zgodnej z grupą krwi dziecka lub w osoczu grupy AB;
3) w konflikcie serologicznym w zakresie innych antygenów niż wymienione w pkt 1 i 2, w szczególności w antygenie K, c, E, Fya, krew do przetoczenia nie może zawierać antygenu odpowiedzialnego za uodpornienie matki.
2. W okresie pierwszych 4 miesięcy życia dziecka do następnych przetoczeń stosuje się krew tej samej grupy ABO i RhD, jak do pierwszej transfuzji.
Rozdział 5
Czuwanie nad bezpieczeństwem krwi
§ 41. [Czuwanie nad bezpieczeństwem krwi]
1) ciężkich niepożądanych zdarzeń;
2) ciężkich powikłań u dawców lub biorców;
3) kontroli epidemiologicznej dawców.
2. Do centrum należy zgłaszać każde ciężkie powikłanie obserwowane u biorcy krwi podczas przetoczenia lub po jego zakończeniu, które można przypisać jakości lub bezpieczeństwu krwi i jej składników.
3. Powikłania poprzetoczeniowe określone w § 14 ust. 1 i 2 zgłasza się na formularzu zgłoszenia powikłania poprzetoczeniowego.
4. Bank krwi, pracownia serologii lub immunologii transfuzjologicznej i podmiot leczniczy zgłaszają do centrum wszelkie niepożądane zdarzenia (wypadki lub błędy) związane z pobieraniem próbek, badaniem, przechowywaniem oraz wydaniem krwi i jej składników, wpływające na ich jakość i bezpieczeństwo oraz wszelkie niepożądane zdarzenia związane z przeprowadzaniem zabiegu przetoczenia.
5. W ramach czuwania nad bezpieczeństwem krwi do centrum przesyła się raporty dotyczące wczesnych i opóźnionych powikłań poprzetoczeniowych, zakażeń biologicznymi czynnikami chorobotwórczymi przenoszonymi drogą krwi oraz wystąpienia alloimmunizacji antygenami krwinek czerwonych, HLA, granulocytów i płytek krwi.
6. Lekarz odpowiedzialny za przetoczenie rejestruje błędy, które mogły zagrażać bezpieczeństwu pacjenta, ale były usunięte przed przetoczeniem (zdarzenia bliskie celu), i raporty o nich przesyła do centrum.
7. Jeżeli aktualne badanie dawcy wykazało potwierdzoną obecność zakażenia wirusami HBV, HCV lub HIV, centrum rozpoczyna procedurę „spojrzenie wstecz” w celu ustalenia biorców krwi, którzy w okresie „okienka diagnostycznego” u dawcy mogli ulec zakażeniu tymi wirusami.
8. Lekarz, który sprawował opiekę nad pacjentem, u którego wystąpiło podejrzenie zakażenia jednym z wirusów HBV, HCV, HIV, lub lekarz wyznaczony przez kierownika jednostki lub komórki organizacyjnej przedsiębiorstwa podmiotu leczniczego, ma obowiązek poinformować o tym pacjenta i zlecić odpowiednie badania w celu potwierdzenia lub wykluczenia zakażenia. Podmiot leczniczy informuje centrum o rezultatach przeprowadzonej procedury, również w przypadku gdy badania nie zostały wykonane wraz z podaniem przyczyny niewykonania badań.
9. Jeżeli u biorcy rozpoznano TRALI, centrum rozpoczyna procedurę „śledzenia wstecz” w celu stwierdzenia, czy krew i jej składniki od tego samego dawcy spowodowały wymienione powikłania u innych biorców.
Rozdział 6
Przepisy przejściowe i przepis końcowy
§ 42. [Wykształcenie osoby zatrudnionej na stanowisku kierownika pracowni serologii lub immunologii transfuzjologicznej]
1) osoba zatrudniona na tym stanowisku w dniu wejścia w życie rozporządzenia nieposiadająca kwalifikacji, o których mowa w § 28 ust. 1 i 2, lub
2) diagnosta laboratoryjny posiadający co najmniej specjalizację I stopnia z diagnostyki laboratoryjnej lub analityki klinicznej oraz co najmniej 3-letnie doświadczenie zawodowe w wykonywaniu badań serologicznych
– pod warunkiem, że osoba ta posiada zaświadczenie uprawniające do wykonywania badań i autoryzacji wyników.
Minister Zdrowia: B.A. Arłukowicz
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 18 listopada 2011 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 248, poz. 1495 i Nr 284, poz. 1672).
2) Niniejsze rozporządzenie dokonuje w zakresie swojej regulacji wdrożenia:
1) dyrektywy 2002/98/WE Parlamentu Europejskiego i Rady z dnia 27 stycznia 2003 r. ustanawiającej normy jakości i bezpiecznego pobierania, badania, preparatyki, przechowywania i wydawania krwi ludzkiej i składników krwi oraz zmieniającej dyrektywę 2001/83/WE (Dz. Urz. UE L 33 z 08.02.2003, str. 30, z późn. zm.; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 15, t. 7, str. 346, z późn. zm.);
2) dyrektywy Komisji 2004/33/WE z dnia 22 marca 2004 r. wykonującej dyrektywę 2002/98/WE Parlamentu Europejskiego i Rady w zakresie niektórych wymagań technicznych dotyczących krwi i składników krwi (Dz. Urz. UE L 91 z 30.03.2004, str. 25, z późn. zm.; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 15, t. 8, str. 272, z późn. zm.);
3) dyrektywy Komisji 2005/61/WE z dnia 30 września 2005 r. wykonującej dyrektywę 2002/98/WE Parlamentu Europejskiego i Rady w zakresie wymogów dotyczących śledzenia losów krwi oraz powiadamiania o poważnych, niepożądanych reakcjach i zdarzeniach (Dz. Urz. UE L 256 z 01.10.2005, str. 32);
4) dyrektywy Komisji 2005/62/WE z dnia 30 września 2005 r. wykonującej dyrektywę 2002/98/WE Parlamentu Europejskiego i Rady w zakresie norm i specyfikacji wspólnotowych odnoszących się do systemu jakości obowiązującego w placówkach służby krwi (Dz. Urz. UE L 256 z 01.10.2005, str. 41).
3) Zmiany wymienionej ustawy zostały ogłoszone w Dz. U. z 1998 r. Nr 117, poz. 756, z 2001 r. Nr 126, poz. 1382, z 2003 r. Nr 223, poz. 2215, z 2007 r. Nr 166, poz. 1172, z 2010 r. Nr 96, poz. 620, z 2011 r. Nr 112, poz. 654 oraz z 2012 r. poz. 742 i 908.
4) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2005 r. Nr 119, poz. 1015, z 2006 r. Nr 117, poz. 790, z 2009 r. Nr 76, poz. 641 oraz z 2011 r. Nr 112, poz. 654 i Nr 113, poz. 657.
5) Niniejsze rozporządzenie było poprzedzone rozporządzeniem Ministra Zdrowia z dnia 19 września 2005 r. w sprawie określenia sposobu i organizacji leczenia krwią w zakładach opieki zdrowotnej, w których przebywają pacjenci ze wskazaniami do leczenia krwią i jej składnikami (Dz. U. Nr 191, poz. 1607 oraz z 2010 r. Nr 159, poz. 1072).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 11 grudnia 2012 r. (poz. 5)
Załącznik nr 1
WZÓR* – KSIĄŻKA TRANSFUZYJNA
(w poziomym układzie strony)
Strona 1
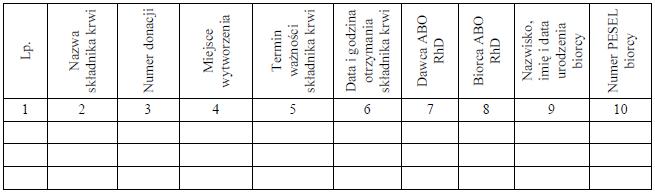
Strona 2

* Układ graficzny nieobowiązujący.
Załącznik nr 2
WZÓR* – STANDARDOWA PROCEDURA OPERACYJNA (SOP)
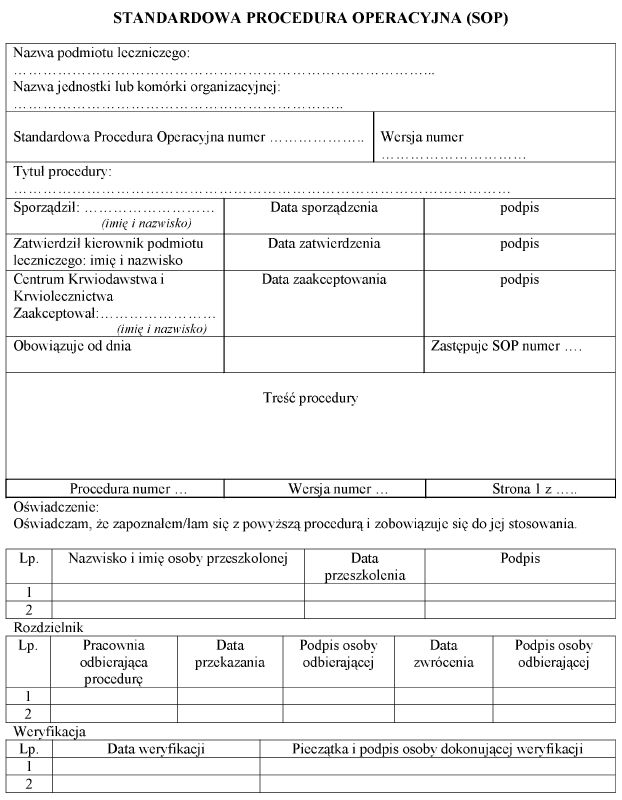
* Układ graficzny nieobowiązujący.
Załącznik nr 3
WZÓR* – KARTA IDENTYFIKACYJNA GRUPY KRWI
Strona 1

Strona 2

* Układ graficzny nieobowiązujący.
Załącznik nr 4
WZÓR* – ZLECENIE NA BADANIE GRUPY KRWI

* Układ graficzny nieobowiązujący.
Załącznik nr 5
WZÓR – ZAMÓWIENIE INDYWIDUALNE NA KREW I JEJ SKŁADNIKI

* Układ graficzny nieobowiązujący.
Załącznik nr 6
WZÓR* – ZLECENIE NA WYKONANIE PRÓBY ZGODNOŚCI

* Układ graficzny nieobowiązujący.
Załącznik nr 7
WZÓR* – ZLECENIE NA KREW DO PILNEJ TRANSFUZJI

* Układ graficzny nieobowiązujący.
Załącznik nr 8
WZÓR* – WYNIK PRÓBY ZGODNOŚCI
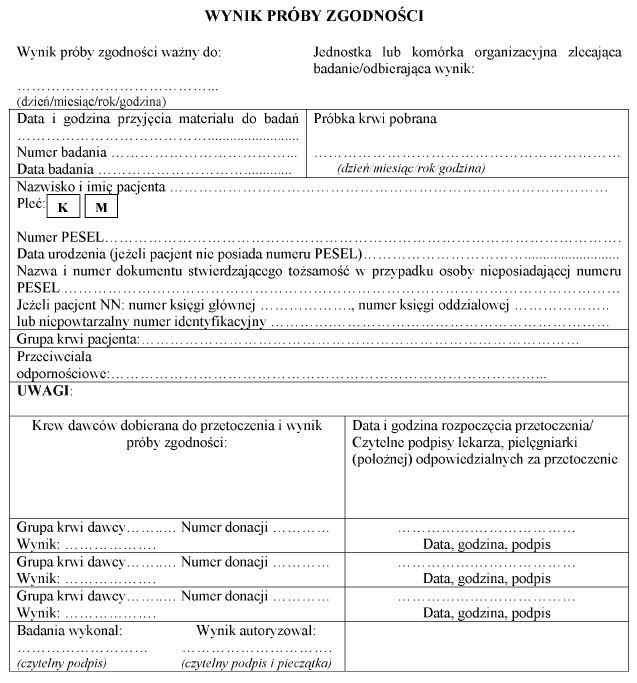
* Układ graficzny nieobowiązujący.
Załącznik nr 9
WZÓR – ZGŁOSZENIE POWIKŁANIA POPRZETOCZENIOWEGO
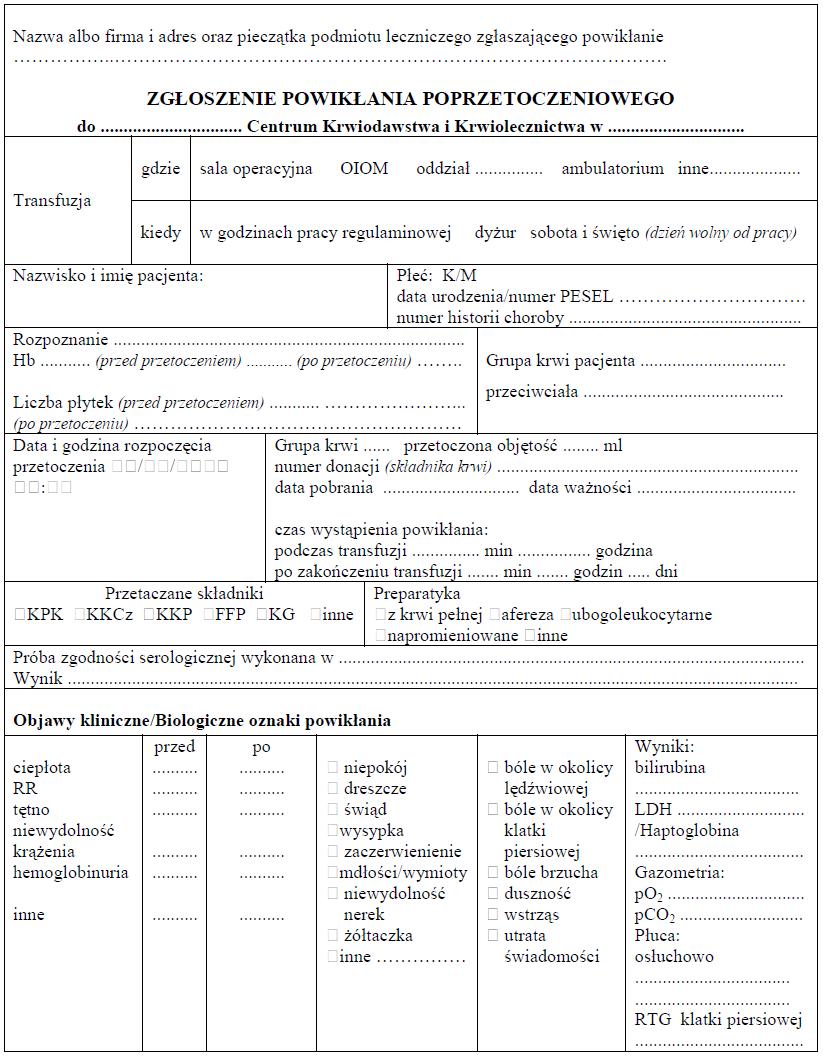
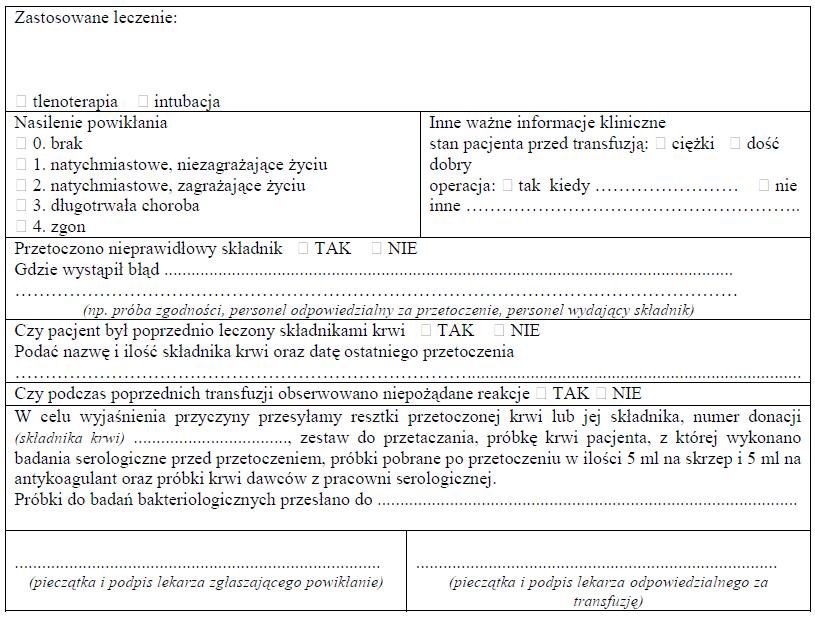
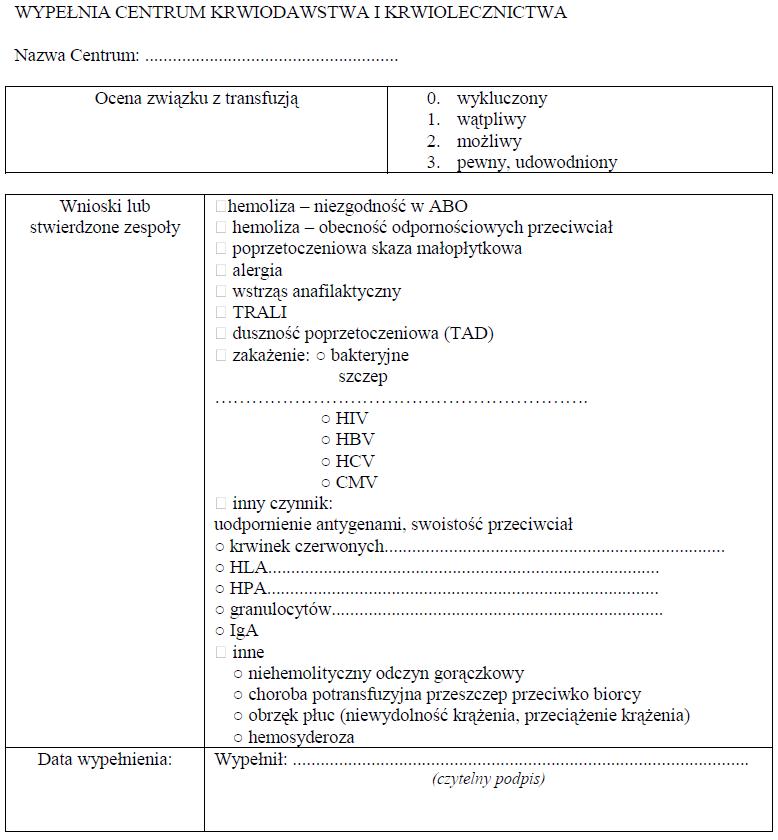
Załącznik nr 10
WZÓR* – ZAMÓWIENIE ZBIORCZE NA KREW LUB JEJ SKŁADNIKI

* Układ graficzny nieobowiązujący.
Załącznik nr 11
WZÓR* – KSIĄŻKA BADAŃ GRUP KRWI
(w poziomym układzie strony)
Strona 1
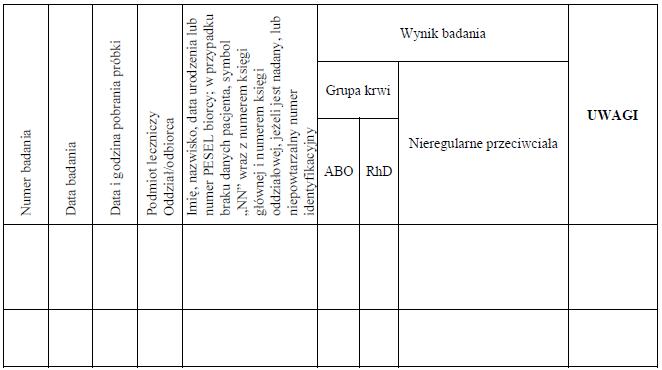
Strona 2
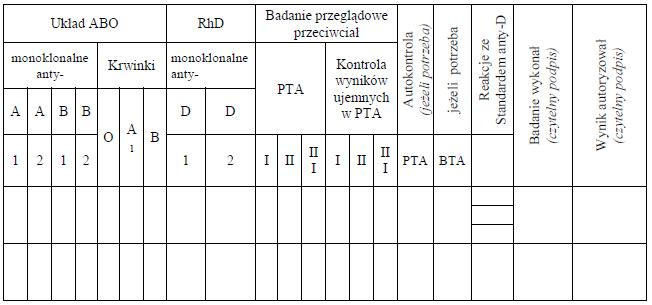
* Układ graficzny nieobowiązujący.
Załącznik nr 12
WZÓR* – KSIĄŻKA PRÓB ZGODNOŚCI
(w poziomym układzie strony)
Strona 1
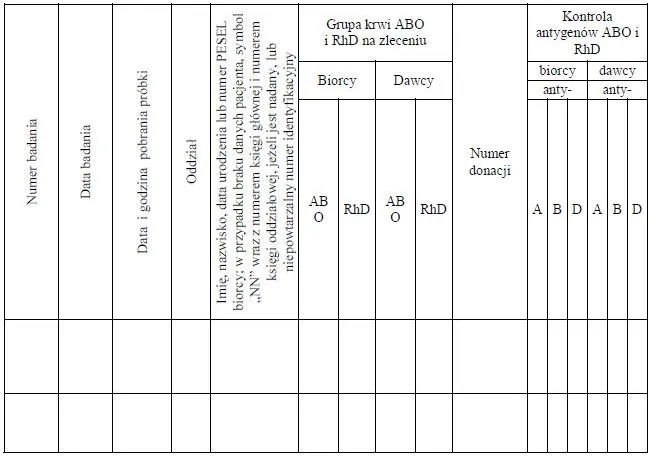
Strona 2
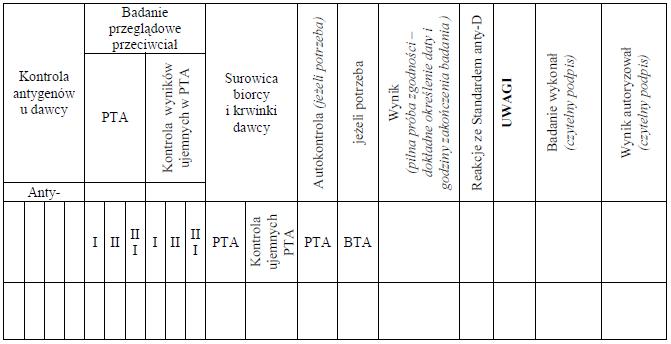
* Układ graficzny nieobowiązujący.
Załącznik nr 13
WZÓR* – ZLECENIE NA KONSULTACYJNE BADANIE IMMUNOHEMATOLOGICZNE
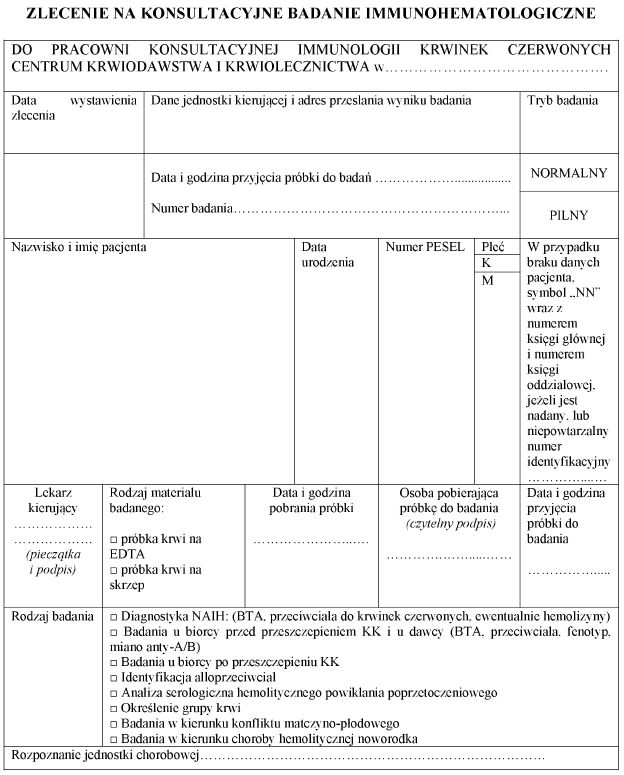

* Układ graficzny nieobowiązujący.
Załącznik nr 14
WZÓR – ZLECENIE NA WYKONANIE BADAŃ IMMUNOHEMATOLOGICZNYCH KWALIFIKUJĄCYCH DO PODANIA IMMUNOGLOBINY ANTY-D

Załącznik nr 15
WZÓR* – WYNIK BADAŃ IMMUNOHEMATOLOGICZNYCH KWALIFIKUJĄCYCH DO PODANIA IMMUNOGLOBINY ANTY-D

* Układ graficzny nieobowiązujący.
Załącznik nr 16
WZÓR* – WYNIK BADANIA GRUPY KRWI

- Data ogłoszenia: 2013-01-04
- Data wejścia w życie: 2013-01-19
- Data obowiązywania: 2013-01-19
- Dokument traci ważność: 2017-09-12

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA