REKLAMA
Dziennik Ustaw - rok 2011 nr 33 poz. 167
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 2 lutego 2011 r.
w sprawie kryteriów raportowania zdarzeń z wyrobami, sposobu zgłaszania incydentów medycznych i działań z zakresu bezpieczeństwa wyrobów
Na podstawie art. 85 ustawy z dnia 20 maja 2010 r. o wyrobach medycznych (Dz. U. Nr 107, poz. 679) zarządza się, co następuje:
1) kryteria raportowania zdarzeń z wyrobami i incydentów medycznych;
2) wzór formularza zgłoszenia incydentu medycznego;
3) wzór formularza raportu wytwórcy o incydencie medycznym;
4) wzór formularza raportu o FSCA;
5) wzór notatki bezpieczeństwa;
6) wzór formularza NCAR;
7) sposób przesyłania zgłoszeń, raportów i notatek, o których mowa w pkt 2–6;
8) szczegółowy tryb postępowania podmiotów biorących udział w działaniach dotyczących incydentu medycznego, FSCA i innych działaniach z zakresu bezpieczeństwa wyrobów.
1) wystąpiło zdarzenie lub stwierdzono sytuację, która może powodować zagrożenie dla pacjenta lub użytkownika, w szczególności gdy wyniki badań wyrobu, analiza informacji dostarczonych wraz z wyrobem lub informacje naukowe wskazują czynnik, który może doprowadzić do takiego zagrożenia;
2) istnieje uzasadnione podejrzenie, że zdarzenie lub sytuację mógł lub może spowodować wyrób;
3) doprowadziło, mogło lub może doprowadzić do śmierci lub poważnego pogorszenia stanu zdrowia pacjenta, użytkownika lub w przypadku wyrobu medycznego do diagnostyki in vitro – pośrednio także innej osoby.
2. Obowiązek raportowania nie dotyczy zdarzeń z wyrobem takich jak:
1) niezdatność wyrobu, która jest zawsze możliwa do stwierdzenia i która nie mogłaby być niewykryta przez użytkownika przed użyciem wyrobu;
2) zdarzenie wynikające ze stanu, w którym pacjent znajdował się przed użyciem wyrobu lub w którym znalazł się w trakcie stosowania wyrobu, o ile wytwórca powziął informację, że wyrób działał, tak jak powinien, i nie spowodował, ani nie przyczynił się do śmierci lub poważnego pogorszenia stanu zdrowia;
3) zdarzenie wynikające z przekroczenia terminu ważności, czasu lub krotności bezpiecznego używania albo terminu okresowego przeglądu lub obsługi serwisowej, podanych w oznakowaniu lub instrukcji używania wyrobu, jeżeli rodzaj niezdatności jest znany lub spodziewany;
4) zdarzenie, które nie doprowadziło do śmierci lub poważnego pogorszenia stanu zdrowia pacjenta, ponieważ zadziałały, zgodnie z zastosowaną normą lub udokumentowanymi danymi wejściowymi do projektowania, cechy konstrukcyjne chroniące przed niezdatnością stwarzającą zagrożenie, o ile pacjent nie był narażony na niebezpieczeństwo oraz typ alarmu zastosowanego jako środek ochronny jest powszechnie uznany w przypadku danego rodzaju wyrobów;
5) działania niepożądane, rozumiane jako każde niekorzystne i niezamierzone działanie wyrobu, które spełniają łącznie następujące warunki:
a) są wyraźnie określone w oznakowaniu lub instrukcji używania wyrobu,
b) są dobrze znane z piśmiennictwa naukowego, badań klinicznych lub praktyki klinicznej jako przewidywalne, oraz określono, jakościowo lub ilościowo, prawdopodobieństwo ich wystąpienia podczas zgodnego z przeznaczeniem użycia i działania wyrobu,
c) zanim wystąpiły, zostały opisane w dokumentacji wyrobu, w tym oszacowano ich ryzyko,
d) są klinicznie akceptowalne, gdyż korzyści z zastosowania danego wyrobu u pacjenta przeważają nad ryzykiem związanym z działaniami niepożądanymi;
6) zdarzenie, które nie doprowadziło do śmierci lub poważnego pogorszenia stanu zdrowia i w przypadku którego oceniono, że ryzyko śmierci lub poważnego pogorszenia stanu zdrowia jest bardzo małe oraz, jeżeli takie ryzyko w ocenie ryzyka zostało określone i udokumentowane jako klinicznie akceptowalne.
3. W przypadku wątpliwości, czy zdarzenie spełnia kryteria raportowania, należy uznać, że spełnia takie kryteria.
2. Opis FSCA – informacje podstawowe oraz przyczyny podjęcia FSCA – część 7 załącznika nr 3 do rozporządzenia – obejmuje w szczególności:
1) odpowiednie części przeprowadzonej analizy ryzyka;
2) opis braków lub wadliwego działania wyrobu;
3) wyjaśnienie potencjalnego zagrożenia związanego z dalszym używaniem wyrobu i ryzyka, jakie stwarza dla pacjenta, użytkownika i innych osób, oraz możliwego ryzyka dla pacjentów związanego z wcześniejszym użyciem wyrobu;
4) wyjaśnienie, dlaczego pozostałe wyroby są bezpieczne, jeżeli działania dotyczą nie wszystkich wyrobów, a jedynie określonych ich serii lub partii.
3. Opis FSCA – informacje o działaniach, które powinien podjąć dystrybutor i użytkownik – część 7 załącznika nr 3 do rozporządzenia – obejmuje zalecenia dotyczące:
1) identyfikacji i kwarantanny wyrobów;
2) metody zwracania, niszczenia lub modyfikacji wyrobów;
3) postępowania z pacjentami;
4) wskazania konieczności:
a) dostarczenia notatki bezpieczeństwa wszystkim osobom, które powinny ją otrzymać,
b) stosowania się do zaleceń określonych w notatce bezpieczeństwa przez wskazany okres,
c) przesłania wytwórcy informacji o wszystkich przekazanych do innych instytucji wyrobach, których dotyczy FSCA, oraz o konieczności dostarczenia tym instytucjom kopii notatki bezpieczeństwa.
2. Prezes Urzędu przesyła formularz NCAR pocztą elektroniczną.
2. W przypadku gdy w terminie, w którym wytwórca lub autoryzowany przedstawiciel jest obowiązany przesłać Raport Wstępny, znane są już wyniki postępowania wyjaśniającego oraz decyzje wytwórcy o przewidywanych lub podjętych działaniach albo o ich braku, Raport Końcowy może być przesłany łącznie z Raportem Wstępnym, na jednym formularzu raportu wytwórcy o incydencie medycznym, w którym zaznacza się rodzaj raportu „Raport Wstępny łącznie z Raportem Końcowym”.
3. Jeżeli w przypadku błędów użytkowych, które nie doprowadziły do śmierci lub poważnego pogorszenia stanu zdrowia pacjenta lub użytkownika, wytwórca lub autoryzowany przedstawiciel:
1) stwierdził znaczący wzrost liczby lub częstości występowania tych błędów lub stwierdził, że mogą one doprowadzić do śmierci lub poważnego pogorszenia stanu zdrowia, lub
2) podjął działania korygujące, aby zapobiec spowodowaniu przez błędy użytkowe śmierci lub poważnego pogorszenia stanu zdrowia pacjenta lub użytkownika
– sytuację kwalifikuje jako incydent medyczny i przesyła Prezesowi Urzędu raport na ten temat.
Minister Zdrowia: E. Kopacz
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 16 listopada 2007 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 216, poz. 1607).
2) Niniejsze rozporządzenie było poprzedzone rozporządzeniem Ministra Zdrowia z dnia 30 kwietnia 2004 r. w sprawie sposobu zgłaszania incydentów medycznych oraz dalszego postępowania po ich zgłoszeniu (Dz. U. Nr 125, poz. 1316).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 2 lutego 2011 r. (poz. 167)
Załącznik nr 1
WZÓR – Formularz zgłoszenia incydentu medycznego

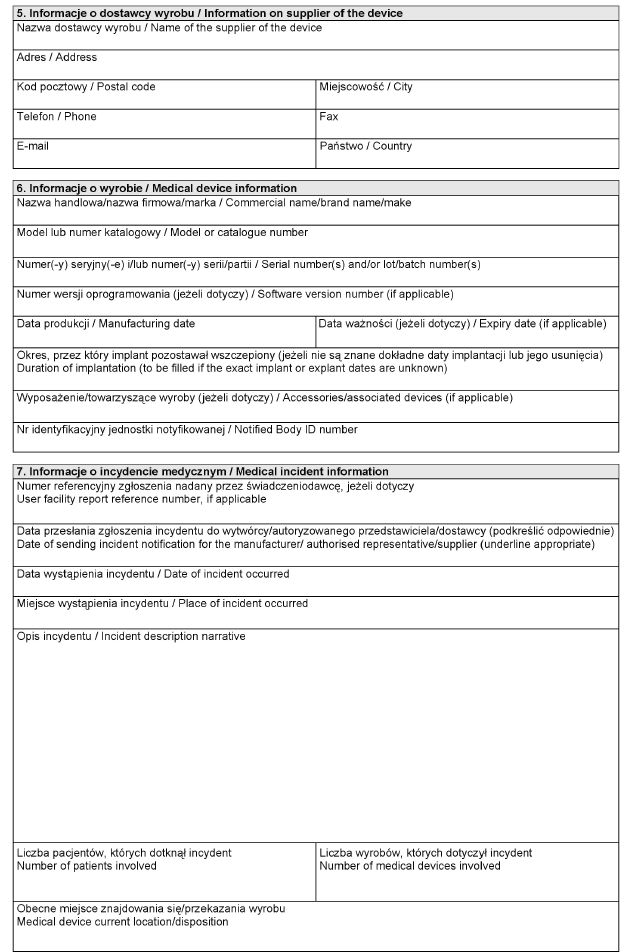

Załącznik nr 2
WZÓR – Formularz raportu wytwórcy o incydencie medycznym


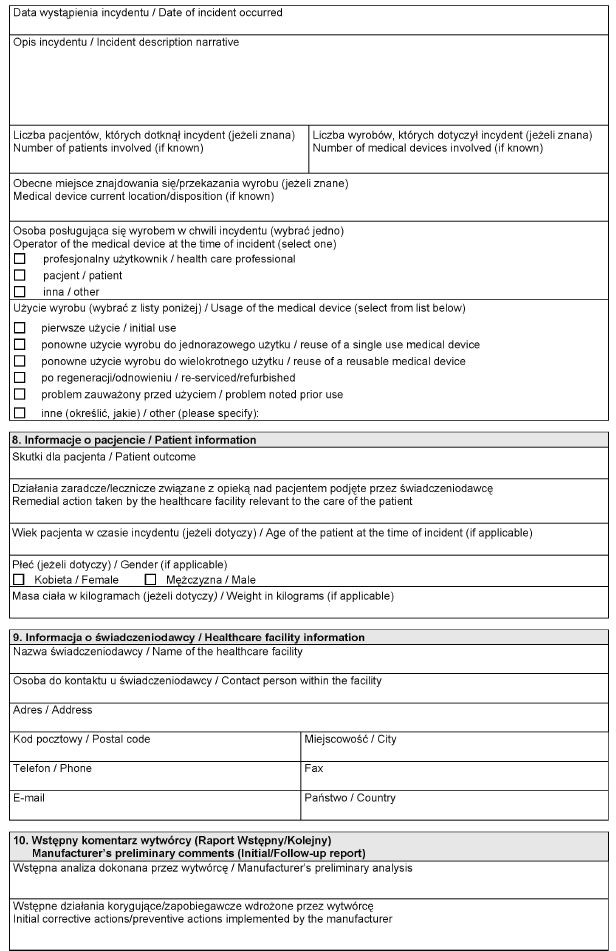
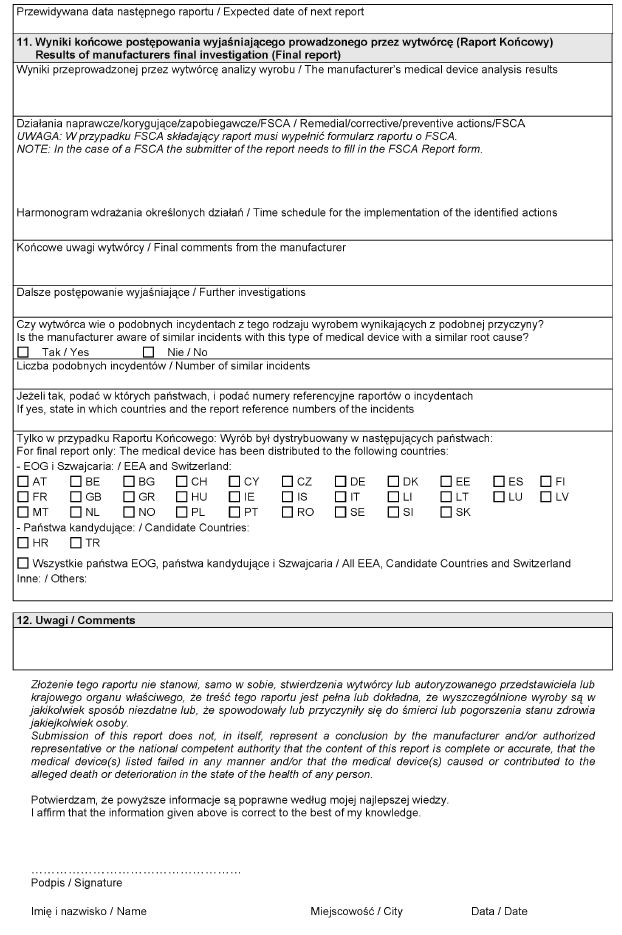
Załącznik nr 3
WZÓR – Formularz raportu o FSCA



Załącznik nr 4
WZÓR – Notatka bezpieczeństwa


Załącznik nr 5
WZÓR – FORMULARZ NCAR

- Data ogłoszenia: 2011-02-16
- Data wejścia w życie: 2011-02-16
- Data obowiązywania: 2011-02-16
- Dokument traci ważność: 2016-02-20

REKLAMA