REKLAMA
Dziennik Ustaw - rok 2010 nr 222 poz. 1453
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 15 listopada 2010 r.
w sprawie wzorów wniosków przedkładanych w związku z badaniem klinicznym, wysokości opłat za złożenie wniosków oraz sprawozdania końcowego z wykonania badania klinicznego
Na podstawie art. 50 ustawy z dnia 20 maja 2010 r. o wyrobach medycznych (Dz. U. Nr 107, poz. 679) zarządza się, co następuje:
1) wzór wniosku o wydanie pozwolenia na prowadzenie badania klinicznego i o wydanie opinii przez komisję bioetyczną o badaniu klinicznym;
2) wzór wniosku o wydanie pozwolenia na wprowadzenie zmian w badaniu klinicznym i o wydanie opinii przez komisję bioetyczną o wnioskowanych zmianach w badaniu klinicznym;
3) wysokość opłaty za złożenie wniosku o wydanie pozwolenia na prowadzenie badania klinicznego;
4) wysokość opłaty za złożenie wniosku o wydanie pozwolenia na wprowadzenie zmian w badaniu klinicznym;
5) informacje, jakie powinno zawierać sprawozdanie końcowe z wykonania badania klinicznego, o którym mowa w art. 54 ust. 4 ustawy z dnia 20 maja 2010 r. o wyrobach medycznych.
2. Wzór wniosku o wydanie pozwolenia na wprowadzenie zmian w badaniu klinicznym i o wydanie opinii przez komisję bioetyczną o wnioskowanych zmianach w badaniu klinicznym jest określony w załączniku nr 2 do rozporządzenia.
1) na prowadzenie badania klinicznego – 5 000 PLN;
2) na wprowadzenie zmian w badaniu klinicznym – 1 500 PLN.
1) streszczenia prezentującego podstawowe informacje o badaniu klinicznym, w tym:
a) tytuł badania klinicznego,
b) identyfikację badanego wyrobu medycznego albo aktywnego wyrobu medycznego do implantacji, zwanych dalej „wyrobem” – nazwy, typy, modele, nadane im numery,
c) nazwę i adres sponsora oraz autoryzowanego przedstawiciela, jeżeli dotyczy,
d) zestawienie aktów prawnych i norm zharmonizowanych, zgodnie z którymi badanie kliniczne było prowadzone,
e) przedmiot i cele badania klinicznego,
f) informacje dotyczące uczestników badania klinicznego,
g) metodologię badania klinicznego,
h) daty rozpoczęcia i zakończenia badania klinicznego albo datę i przyczynę wstrzymania badania klinicznego, jeżeli miało miejsce,
i) wyniki badania klinicznego,
j) wnioski z badania klinicznego,
k) datę sporządzenia sprawozdania końcowego oraz nazwiska i podpisy jego autorów;
2) wprowadzenia zawierającego uzasadnienie przeprowadzenia badania klinicznego, jego założenia, opis badanej populacji, czas trwania badania klinicznego, mierniki, a także podstawy opracowania protokołu badania klinicznego;
3) opisu materiału i metod:
a) dane wyrobu,
b) opis badanego wyrobu i jego przewidziane zastosowanie oraz opis wprowadzonych modyfikacji wyrobu, jakie miały miejsce w trakcie prowadzonego badania klinicznego,
c) streszczenie protokołu badania klinicznego wraz z opisem wszystkich zmian, jakie miały miejsce w trakcie prowadzonego badania klinicznego, w tym informacje dotyczące:
– celów badania klinicznego,
– projektu badania klinicznego (typ, punkty końcowe, względy etyczne),
– populacji uczestników badania klinicznego (kryteriów włączania i wyłączania, liczebności),
– czasu trwania badania klinicznego,
– leczenia towarzyszącego i stosowanych produktów leczniczych,
– zmiennych losowych przyjętych w badaniu klinicznym,
– analizy statystycznej (hipoteza badawcza, poziomy istotności, kryteria przyjęcia, kryteria odrzucenia wyników, liczebność próby, metody analizy);
4) zestawienia wyników obejmującego:
a) datę rozpoczęcia i zakończenia lub wstrzymania badania klinicznego,
b) liczbę i charakterystykę demograficzną uczestników badania,
c) liczbę użytych wyrobów w odniesieniu do uczestników badania klinicznego oraz ośrodków, jeżeli jest to istotne,
d) potwierdzenie zgodności z protokołem badania klinicznego,
e) analizę bezpieczeństwa zawierającą:
– ocenę bezpieczeństwa stosowania wyrobu,
– zestawienie zdarzeń niepożądanych związanych z badaniem klinicznym wyrobu,
– oszacowanie wagi i konsekwencji zdarzeń niepożądanych, w tym niezbędnej terapii i dodatkowych badań diagnostycznych, laboratoryjnych i podobnych,
– ocenę związku zdarzeń niepożądanych ze stosowanym w badaniu klinicznym wyrobem i z zastosowaną procedurą,
f) analizę działania i skuteczności badanego wyrobu,
g) analizy cząstkowe odniesione do wydzielonych podgrup uczestników, wyrobów, przyrządów, jeżeli mają zastosowanie,
h) analizę wyników badania klinicznego pozwalającą stwierdzić, że:
– kryteria przyjęcia albo odrzucenia osoby z uczestnictwa w badaniu klinicznym spełniła określona część kandydatów w stanie zdrowia uzasadniającym kwalifikację na uczestnika badania klinicznego,
– wyniki dowodzą zgodności z określonymi wymaganiami zasadniczymi,
– informacje dołączone do wyrobu odpowiadają danym klinicznym i danym zebranym przed badaniem klinicznym,
– ryzyko związane z użyciem wyrobu jest akceptowalne w świetle korzyści, jakie mógł odnieść uczestnik badania klinicznego,
i) omówienie dalszego postępowania z danymi odrzuconymi;
5) omówienia (dyskusji) oraz wniosków obejmujących:
a) działanie i bezpieczeństwo wyrobu,
b) stosunek ryzyka do korzyści,
c) wagę i istotność kliniczną wyników w świetle innych istniejących danych,
d) szczególne korzyści oraz specjalne środki ostrożności w stosunku do pojedynczych uczestników badania lub grup ryzyka,
e) wskazania do dalszych badań, w tym badań klinicznych lub zmian konstrukcyjnych wyrobu;
6) omówienia zagadnień etycznych.
1) protokół badania klinicznego wraz z wprowadzonymi zmianami;
2) instrukcję używania badanego wyrobu;
3) tabelaryczne zestawienie badaczy klinicznych w poszczególnych ośrodkach badawczych zawierające imiona i nazwiska badaczy klinicznych wraz z informacjami o ich stopniu naukowym i specjalizacji;
4) wykaz wszystkich pozostałych ośrodków uczestniczących w badaniu klinicznym, w tym laboratoriów, konsultantów oraz ośrodków badawczych, innych niż wymienione w pkt 3;
5) wykaz osób monitorujących badanie kliniczne;
6) wykaz statystyków (planowanie i analiza wyników badania klinicznego), jeżeli ma to zastosowanie;
7) wykaz i opinie komisji bioetycznych;
8) opinie niezależnych ekspertów, jeżeli ma to zastosowanie;
9) tabelaryczne zestawienie zdarzeń i działań niepożądanych, jeżeli miały miejsce.
Minister Zdrowia: E. Kopacz
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 16 listopada 2007 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 216, poz. 1607).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 15 listopada 2010 r. (poz. 1453)
Załącznik nr 1
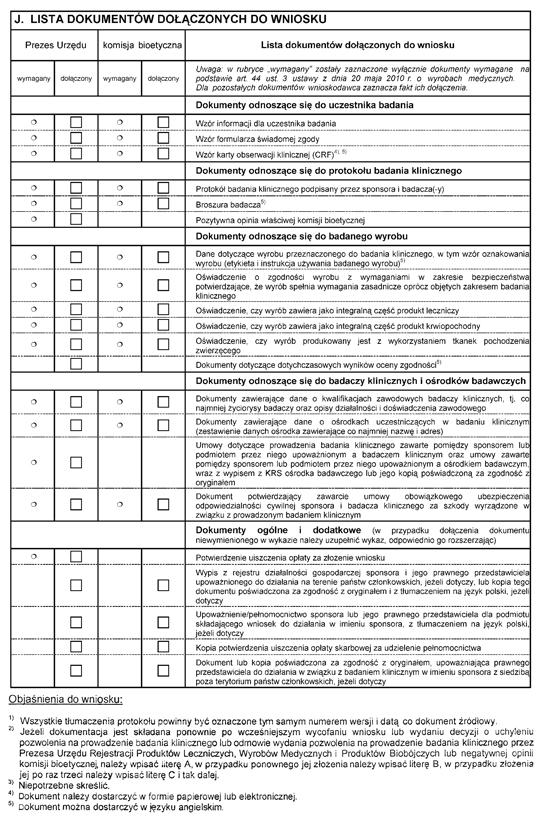
WNIOSEK O WYDANIE POZWOLENIA NA PROWADZENIE BADANIA KLINICZNEGO /
WNIOSEK O WYDANIE OPINII PRZEZ KOMISJĘ BIOETYCZNĄ O BADANIU KLINICZNYM






Załącznik nr 2
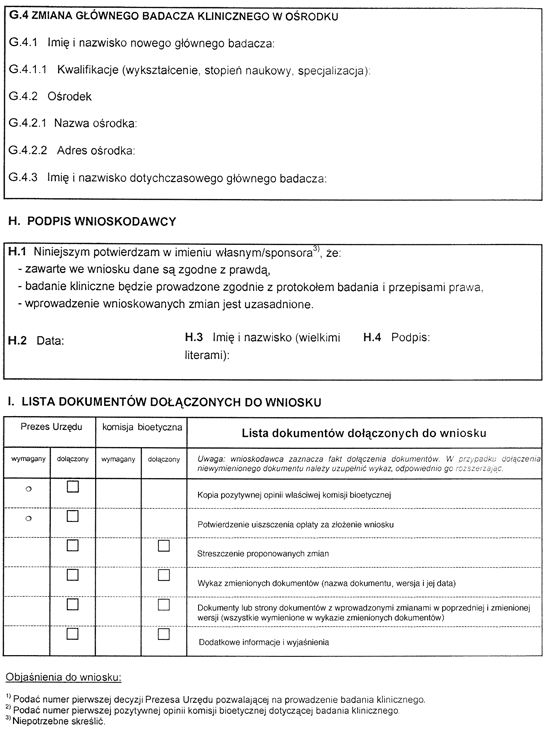
WNIOSEK O WYDANIE POZWOLENIA NA WPROWADZENIE ZMIAN W BADANIU KLINICZNYM /
WNIOSEK O WYDANIE OPINII PRZEZ KOMISJĘ BIOETYCZNĄ O WNIOSKOWANYCH ZMIANACH W BADANIU KLINICZNYM



- Data ogłoszenia: 2010-11-25
- Data wejścia w życie: 2010-11-25
- Data obowiązywania: 2010-11-25
- Dokument traci ważność: 2016-02-20

REKLAMA