REKLAMA
Dziennik Ustaw - rok 2009 nr 39 poz. 321
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 20 lutego 2009 r.
w sprawie wymagań dotyczących oznakowania opakowań produktu leczniczego i treści ulotki2)
Na podstawie art. 26 ust. 2 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne (Dz. U. z 2008 r. Nr 45, poz. 271, z późn. zm.3)) zarządza się, co następuje:
1) nazwę produktu leczniczego;
2) moc produktu leczniczego, jeżeli produkt jest dostępny w kilku mocach;
3) postać farmaceutyczną, jeżeli produkt jest dostępny w kilku postaciach.
1) z zachowaniem kolejności:
a) nazwę produktu leczniczego i nazwę powszechnie stosowaną substancji czynnej, jeżeli produkt zawiera tylko jedną substancję czynną; jeżeli produkt zawiera dwie lub trzy substancje czynne, podaje się nazwy powszechnie stosowane wszystkich tych substancji oddzielone znakiem „+",
b) moc produktu leczniczego, jeżeli produkt leczniczy zawiera tylko jedną substancję czynną; jeżeli produkt zawiera dwie lub trzy substancje czynne, podaje się moc produktu w przeliczeniu na każdą z tych substancji oddzieloną znakiem „+",
c) określenie postaci farmaceutycznej,
d) informację, czy produkt jest przeznaczony dla niemowląt, dzieci lub dorosłych, jeżeli dotyczy,
e) określenie wielkości opakowania, z podaniem masy, objętości lub liczby jednostek dawkowania produktu leczniczego,
2) zawartość substancji czynnych określoną jakościowo i ilościowo na jednostkę dawkowania, objętości lub masy, z zastosowaniem nazw powszechnie stosowanych,
3) wykaz substancji pomocniczych o uznanym działaniu i skutku, określonych na podstawie art. 65 dyrektywy 2001/83/WE Parlamentu Europejskiego i Rady z dnia 6 listopada 2001 r. w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi (Dz. Urz. WE L 311 z 28.11.2001, str. 67, z późn. zm.; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 27, str. 69, z późn. zm.), a w przypadku produktów leczniczych stosowanych pozajelitowo, miejscowo i do oczu – wykaz wszystkich substancji pomocniczych,
4) sposób stosowania i w razie konieczności drogę podania,
5) ostrzeżenie dotyczące przechowywania w miejscu niedostępnym i niewidocznym dla dzieci,
6) ostrzeżenia dotyczące miejsca oraz sposobu przechowywania, jeżeli występują,
7) inne ostrzeżenia specjalne, jeżeli są konieczne,
8) kategorię dostępności, o której mowa w art. 23a ust. 1 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, zwanej dalej „ustawą",
9) termin ważności (miesiąc i rok), a dla leków gotowych, które wymagają specjalnego postępowania bezpośrednio przed ich zastosowaniem – także, jeżeli jest to niezbędne ze względu na właściwości produktu leczniczego, oznaczenie okresu przydatności po doprowadzeniu leku do postaci nadającej się do zastosowania,
10) specjalne środki ostrożności, które należy zachować podczas usuwania niezużytego produktu leczniczego lub odpadów pochodzących z produktów leczniczych, jeżeli występują,
11) nazwę i adres podmiotu odpowiedzialnego oraz, jeżeli dotyczy, nazwę i adres przedstawiciela podmiotu odpowiedzialnego,
12) numer pozwolenia na dopuszczenie do obrotu,
13) numer serii,
14) instrukcję użycia, jeżeli produkt leczniczy jest wydawany bez przepisu lekarza,
15) kod kreskowy EAN UCC,
16) w przypadku tradycyjnego produktu leczniczego roślinnego:
a) informację, że jego wskazanie opiera się wyłącznie na długim okresie stosowania,
b) zalecenie, że użytkownik powinien skonsultować się z lekarzem, jeżeli objawy nie ustępują w czasie stosowania produktu lub występują działania niepożądane, niewymienione w ulotce
– z zastrzeżeniem § 9, 10, 11.
2. Oznakowanie produktu leczniczego umieszcza się w sposób trwały na etykiecie przymocowanej do opakowania lub bezpośrednio na opakowaniu.
1) blistrach – zamieszcza się co najmniej następujące informacje:
a) nazwę produktu leczniczego, określoną zgodnie z § 3 ust. 1 pkt 1 lit. a–c,
b) nazwę podmiotu odpowiedzialnego,
c) termin ważności (miesiąc i rok),
d) numer serii,
2) których oznakowanie zgodnie z wymaganiami określonymi w § 3 i 7 nie jest możliwe ze względu na małe rozmiary tych opakowań – zamieszcza się co najmniej następujące informacje:
a) nazwę produktu leczniczego określoną zgodnie z § 3 ust. 1 pkt 1 lit. a–c, a także – w razie konieczności drogę podania,
b) sposób stosowania, jeżeli jest to konieczne,
c) termin ważności (miesiąc i rok),
d) numer serii,
e) zawartość opakowania bezpośredniego, z podaniem masy, objętości lub liczby jednostek dawki produktu leczniczego
– z zastrzeżeniem § 8, 12, 14.
2. Pełna treść ulotki jest udostępniana w formie właściwej dla osób niewidomych i słabowidzących.
1) informacje umożliwiające identyfikację produktu leczniczego:
a) określone zgodnie z § 3 ust. 1 pkt 1,
b) określenie grupy farmakologiczno-terapeutycznej lub sposobu działania – w sposób zrozumiały dla pacjenta,
2) wskazania do stosowania, a w przypadku tradycyjnego produktu leczniczego roślinnego dodatkowo informację, że produkt jest tradycyjnym produktem leczniczym roślinnym do stosowania w określonych wskazaniach, wynikających wyłącznie z jego długotrwałego stosowania,
3) informacje niezbędne przed rozpoczęciem stosowania produktu leczniczego:
a) przeciwwskazania,
b) odpowiednie środki ostrożności związane ze stosowaniem, a w przypadku tradycyjnego leczniczego produktu roślinnego informację, że użytkownik powinien skonsultować się z lekarzem, jeżeli objawy nie ustępują w czasie stosowania produktu lub występują działania niepożądane, niewymienione w ulotce,
c) interakcje z innymi produktami leczniczymi oraz inne rodzaje interakcji (np. z alkoholem, tytoniem, żywnością) mogące zaburzać działanie produktu leczniczego,
d) ostrzeżenia specjalne dotyczące:
– szczególnych grup użytkowników (w szczególności dzieci, kobiet w ciąży i karmiących piersią, pacjentów w podeszłym wieku, pacjentów z określonymi schorzeniami, takimi jak niewydolność wątroby lub nerek), jeżeli jest to konieczne,
– wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn,
– substancji pomocniczych, o których mowa w § 3 ust. 1 pkt 3, mających istotne znaczenie dla właściwego stosowania produktu leczniczego,
4) informacje niezbędne do prawidłowego stosowania produktu leczniczego, w szczególności:
a) dawkowanie,
b) sposób stosowania i droga podania,
c) częstość stosowania z określeniem pory dnia, jeżeli to konieczne,
d) czas trwania leczenia, jeżeli powinien być ograniczony,
e) objawy i postępowanie w przypadku przedawkowania,
f) postępowanie w przypadku pominięcia dawki leku,
g) ryzyko wystąpienia zespołu z odstawienia, jeżeli to konieczne,
h) zalecenie zasięgnięcia porady lekarza lub farmaceuty w celu uzyskania wyjaśnień co do stosowania produktu leczniczego,
5) opis działań niepożądanych, które mogą wystąpić podczas prawidłowego stosowania produktu leczniczego, oraz, jeżeli jest to konieczne, opis postępowania, które należy podjąć w razie ich wystąpienia, a ponadto wyraźne zalecenie o konieczności informowania lekarza lub farmaceuty o wystąpieniu objawów niepożądanych,
6) w odniesieniu do terminu ważności podanego na opakowaniu następujące informacje:
a) ostrzeżenie o zakazie stosowania produktu leczniczego po upływie terminu ważności,
b) opis szczególnych warunków przechowywania, jeżeli są one wymagane,
c) opis zmian świadczących o pogorszeniu jakości produktu leczniczego, jeżeli to konieczne,
d) skład jakościowy i ilościowy substancji czynnych z zastosowaniem nazw, o których mowa w § 3 ust. 1 pkt 2, oraz skład jakościowy substancji pomocniczych określony zgodnie z § 3 ust. 1 pkt 3,
e) zawartość opakowania z podaniem masy, objętości lub liczby jednostek dawkowania,
f) nazwę i adres podmiotu odpowiedzialnego i jeżeli dotyczy – nazwę przedstawiciela na terytorium Rzeczypospolitej Polskiej,
g) nazwę i adres wytwórcy lub importera, u którego następuje zwolnienie serii,
7) nazwy produktu leczniczego w innych państwach członkowskich Wspólnoty, w przypadku gdy produkt został dopuszczony do obrotu w procedurach europejskich, o ile są różne,
8) datę zatwierdzenia tekstu ulotki
– z zastrzeżeniem § 7, 9, 11, 13, 14.
2. Wszystkie dane umieszczone na opakowaniu zewnętrznym i opakowaniu bezpośrednim muszą być czytelne, zrozumiałe i nieusuwalne.
2. Opakowania, o których mowa w ust. 1, są oznakowane następująco:
1) na etykiecie umieszczonej na osłonie zamieszcza się:
a) informacje szczegółowe określone w § 3 ust. 1,
b) objaśnienia znaczenia symboli użytych na fiolce i liczbę kapsułek, a w przypadku płynów – liczbę mililitrów w pojemniku, a jeżeli jest to konieczne, dla danego (określonego) czasu i określonej daty, ilości radioaktywności w dawce lub fiolce;
2) na fiolce zamieszcza się następujące informacje:
a) nazwę lub symbol produktu leczniczego, włącznie z nazwą lub symbolem chemicznym radionuklidu,
b) numer serii i termin ważności (miesiąc i rok),
c) międzynarodowy symbol radioaktywności,
d) nazwę podmiotu odpowiedzialnego,
e) ilość radioaktywności określoną zgodnie z pkt 1 lit. b.
1) nazwę produktu leczniczego homeopatycznego, o której mowa w art. 21 ust. 2 pkt 3 ustawy;
2) nazwę i adres podmiotu odpowiedzialnego;
3) stopień rozcieńczenia;
4) sposób stosowania i drogę podania;
5) termin ważności (miesiąc i rok);
6) określenie postaci farmaceutycznej i zawartości opakowania przez podanie masy, objętości lub liczby dawek produktu leczniczego homeopatycznego;
7) ostrzeżenia dotyczące przechowywania produktu, jeżeli występują;
8) ostrzeżenia specjalne dotyczące przechowywania w miejscu niedostępnym i niewidocznym dla dzieci;
9) ostrzeżenia specjalne, jeżeli jest to konieczne;
10) numer serii;
11) numer pozwolenia na dopuszczenie do obrotu;
12) określenie „Homeopatyczny produkt leczniczy bez wskazań leczniczych"
13) zalecenie dotyczące konsultacji z lekarzem w przypadku pojawienia się niepożądanych objawów w czasie stosowania produktu leczniczego homeopatycznego;
14) kod kreskowy EAN UCC;
15) kategorię dostępności, o której mowa w art. 23a ust. 1 ustawy.
1) nazwę produktu leczniczego homeopatycznego odpowiednio do danego produktu, o którym mowa w § 10 lub 11;
2) nazwę podmiotu odpowiedzialnego;
3) termin ważności (miesiąc i rok);
4) numer serii.
1) wartość energetyczną wyrażoną w kJ i kcal oraz zawartość białka, węglowodanów i tłuszczów, wyrażoną liczbowo jako zawartość w 100 g lub 100 ml produktu występującego w obrocie i jeżeli to dotyczy – na 100 g lub 100 ml produktu gotowego do użytku, zgodnie z zaleceniami wytwórcy; informacje te mogą ponadto być podane w przeliczeniu na posiłek wielkości wyszczególnionej na opakowaniu lub w przeliczeniu na porcję, pod warunkiem że podano liczbę porcji w opakowaniu;
2) średnią zawartość każdej substancji mineralnej i każdej witaminy obecnej w produkcie, wyrażoną liczbowo jako zawartość w 100 g lub 100 ml produktu występującego w obrocie i jeżeli to dotyczy – na 100 g lub 100 ml produktu gotowego do użytku, zgodnie z zaleceniami wytwórcy; informacje te mogą ponadto być podane w przeliczeniu na posiłek wielkości wyszczególnionej na opakowaniu lub w przeliczeniu na porcję, pod warunkiem że podano liczbę porcji w opakowaniu;
3) osobno zawartość składników białkowych, węglowodanów i tłuszczów lub innych składników odżywczych i ich składników, których podanie jest konieczne do właściwego przewidzianego zastosowania produktu, wyrażoną liczbowo jako zawartość w 100 g lub 100 ml produktu występującego w obrocie i jeżeli to dotyczy – na 100 g lub 100 ml produktu gotowego do użytku, zgodnie z zaleceniami wytwórcy; informacje te mogą ponadto być podane w przeliczeniu na posiłek wielkości wyszczególnionej na opakowaniu lub w przeliczeniu na porcję, pod warunkiem że podano liczbę porcji w opakowaniu;
4) informacje o osmolalności lub osmolarności, jeżeli to dotyczy produktu;
5) informacje o pochodzeniu i rodzaju białek lub hydrolizatów białkowych zawartych w produkcie.
2. Na opakowaniu umieszcza się następujące informacje, poprzedzone słowami „Ważne informacje" lub ich odpowiednikiem:
1) informacja o tym, że produkt musi być stosowany pod nadzorem medycznym;
2) informacja o tym, czy produkt jest odpowiedni do użytku jako wyłączne źródło pożywienia;
3) w razie potrzeby informacja, że produkt jest przewidziany dla określonej grupy wiekowej;
4) w razie potrzeby informacja, że produkt stanowi zagrożenie zdrowia w razie spożycia przez osoby bez chorób, zaburzeń lub stanów medycznych, w których ten produkt jest wskazany.
3. Ponadto na opakowaniu umieszcza się:
1) stwierdzenie „do leczenia dietetycznego ... ", gdzie wolne miejsce należy uzupełnić nazwami chorób, zaburzeń lub stanów medycznych, dla jakich produkt jest wskazany;
2) tam gdzie ma to zastosowanie, informacje dotyczące stosownych środków ostrożności i przeciwwskazań;
3) opis właściwości lub cech charakterystycznych czyniących produkt użytecznym, zwłaszcza odnośnie do substancji odżywczych, których ilość została zwiększona, zmniejszona, które zostały usunięte lub zmienione w inny sposób, a także racjonalne uzasadnienie stosowania produktu;
4) tam gdzie ma to zastosowanie, ostrzeżenie, że produkt nie jest do użytku pozajelitowego.
4. Na opakowaniu umieszcza się także zalecenia dotyczące właściwego przygotowania, stosowania i przechowywania produktu po otwarciu opakowania.
2. Wymagania dotyczące sposobu sporządzania ulotki określa załącznik nr 2 do rozporządzenia.
Minister Zdrowia: E. Kopacz
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 16 listopada 2007 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 216, poz. 1607).
2) Przepisy niniejszego rozporządzenia wdrażają postanowienia dyrektywy 2001/83/WE Parlamentu Europejskiego i Rady z dnia 6 listopada 2001 r. w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi (Dz. Urz. WE L 311 z 28.11.2001, str. 67, z późn. zm.; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 27, str. 69, z późn. zm.).
3) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2008 r. Nr 227, poz. 1505 i Nr 234, poz. 1570 oraz z 2009 r. Nr 18, poz. 97 i Nr 31, poz. 206.
4) Niniejsze rozporządzenie było poprzedzone rozporządzeniem Ministra Zdrowia z dnia 19 grudnia 2002 r. w sprawie wymagań dotyczących oznakowania opakowania produktu leczniczego oraz treści ulotek (Dz. U. Nr 234, poz. 1978), które utraciło moc z dniem 1 listopada 2008 r. na podstawie art. 15 ustawy z dnia 30 marca 2007 r. o zmianie ustawy – Prawo farmaceutyczne oraz o zmianie niektórych innych ustaw (Dz. U. Nr 75, poz. 492).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 20 lutego 2009 r. (poz. 321)
Załącznik nr 1
WYMAGANIA DOTYCZĄCE SPOSOBU SPORZĄDZANIA OZNAKOWANIA OPAKOWAŃ
1. Nazwa produktu leczniczego
Informacje określone w § 3 ust. 1 pkt 1 lit. a rozporządzenia mogą być pisane różną czcionką i zamieszczane w różnych wierszach, ale tak, aby znajdowały się w jednym polu widzenia.
2. Skład jakościowy i ilościowy substancji czynnej
Nazwa substancji czynnej
Nazwa substancji czynnej może być zamieszczona poniżej wiersza z informacją dotyczącą nazwy, mocy i postaci farmaceutycznej, lecz w jednym polu widzenia.
Zawartość substancji czynnej
A. Zawartość substancji czynnej można wyrazić jednym z następujących sposobów:
1) na jednostkę dawkowania;
2) na jednostkę objętości (jeżeli jest to właściwe dla danej postaci farmaceutycznej);
3) na jednostkę masy (jeżeli jest to właściwe dla danej postaci farmaceutycznej).
B. Jeżeli substancja czynna występuje w postaci związku lub pochodnej (np.: soli lub estru), to jej zawartość najlepiej wyrazić w przeliczeniu na ilość aktywnej części cząsteczki, a nie jako ilość całego związku. Jeżeli substancja czynna tworzy sole in situ, to jej zawartość wyraża się w odniesieniu do aktywnej części cząsteczki, po czym podaje się, jaka sól powstaje („tworzy in situ sól...").
C. Różne dawki tej samej substancji określa się w ten sam sposób (niewłaściwe jest użycie w tekście np. 250 mg i 0,25 g), przy czym należy unikać ułamków dziesiętnych, gdyż przy pisaniu tych ułamków przecinek może być łatwo pominięty. Tak więc w zakresie od 1 do 999 mg dawkę należy podawać w miligramach, a nie w gramach. W celu uniknięcia pomyłki mikrogramy zapisuje się całym słowem, a nie skrótem jednostki, chyba że nie ma możliwości zmniejszenia wielkości czcionki do 7 punktów Didota. Dla produktów biologicznych, tam gdzie jest to możliwe, należy stosować jednostki międzynarodowe (j.m.).
D. Produkty lecznicze do podawania pozajelitowego
W informacjach zamieszczonych na opakowaniach jednodawkowych produktów leczniczych do stosowania pozajelitowego zawartość substancji czynnej podaje się w przeliczeniu na 1 ml oraz na całą objętość. W informacjach na opakowaniach wielodawkowych lub o dużych objętościach (np. płyny infuzyjne) zawartość substancji czynnej podaje się odpowiednio w przeliczeniu na 1 ml, na 100 ml lub na 1000 ml. W przypadku produktów leczniczych o dużych objętościach, zawierających sole nieorganiczne, zawartość tych soli podaje się również w milimolach.
1. Zestawy roztworów/emulsji do sporządzania roztworów/emulsji do infuzji znajdujące się w pojemnikach kilkukomorowych (mieszaninę podawaną pacjentowi uzyskuje się przez przerwanie przegród i zmieszanie zawartości komór)
W informacji na opakowaniu podaje się dane dotyczące zawartości poszczególnych substancji czynnych w gotowej mieszaninie:
1) zawartość każdej substancji w całej objętości mieszaniny;
2) stężenie każdej substancji w mieszaninie wyrażone w jednostkach masy na 1 ml, na 100 ml lub na 1000 ml;
3) stężenie molowe jonów nieorganicznych i ewentualnie niektórych jonów organicznych;
4) pH sporządzonej mieszaniny;
5) osmolarność lub osmolalność sporządzonej mieszaniny;
6) ewentualnie dodatkowe informacje ważne dla lekarza, takie jak: całkowita ilość aminokwasów, całkowity azot, wartość energetyczna itp.
2. Koncentraty do stosowania pozajelitowego
W informacji na opakowaniu podaje się:
1) całkowitą zawartość substancji czynnej w opakowaniu;
2) zawartość substancji czynnej w 1 ml koncentratu;
3) zawartość substancji czynnej w 1 ml roztworu otrzymanego po rozcieńczeniu zgodnie z instrukcją, chyba że istnieją też inne sposoby rozcieńczania, których rezultatem są inne ostateczne stężenia substancji czynnej;
4) jasne sformułowanie dotyczące sposobu rozcieńczania (np. „rozcieńczyć przed użyciem – patrz ulotka").
3. Proszek do sporządzania roztworu do podawania pozajelitowego
W informacji na opakowaniu podaje się:
1) całkowitą zawartość substancji czynnej w opakowaniu;
2) zawartość substancji czynnej w 1 ml roztworu otrzymanego po rozcieńczeniu zgodnie z instrukcją, chyba że istnieją też inne sposoby rozcieńczania, których rezultatem są inne ostateczne stężenia substancji czynnej;
3) jasne sformułowanie dotyczące sposobu rozcieńczania (np. „rozcieńczyć przed użyciem – patrz ulotka").
4. Rozpuszczalniki przeznaczone do rozcieńczania koncentratu lub rozpuszczania proszku do podawania pozajelitowego
W informacji na opakowaniu podaje się objętość rozpuszczalnika możliwą do pobrania z opakowania.
E. Systemy transdermalne
W informacji na opakowaniu podaje się:
1) zawartość substancji czynnych w jednym plastrze;
2) średnią dawkę dostarczaną pacjentowi w jednostce czasu (np. dawkę wchłoniętą w ciągu godziny, doby);
3) powierzchnię przylegania.
Każdą z tych informacji podaje się w sposób przejrzysty i oddzielnie, tak aby można je było odróżnić od siebie i aby nie mogło nastąpić pomylenie tych informacji podczas wydawania produktu leczniczego.
F. Preparaty wielodawkowe stałe i półpłynne (proszki niedzielone, granulaty, maści i kremy)
Zawartość substancji czynnej określa się w odniesieniu do jednostki dawkowania albo na gram, albo jako procent.
G. Implanty i wkładki wewnątrzmaciczne (zaliczone do produktów leczniczych)
W informacji na opakowaniu podaje się:
1) zawartość substancji czynnych w jednym implancie lub wkładce;
2) średnią dawkę dostarczaną pacjentowi w jednostce czasu (np. dawkę uwolnioną i wchłoniętą w ciągu godziny, doby);
3) całkowity czas (np. godziny, dni), w którym ta średnia dawka będzie dostarczana.
3. Postać farmaceutyczna i jej zawartość w opakowaniu
Określając postać farmaceutyczną, należy korzystać z aktualnego ujednoliconego nazewnictwa Farmakopei Polskiej lub wykazu terminów standardowych Farmakopei Europejskiej. Lista ta zawiera również skrócone nazwy niektórych postaci farmaceutycznych, jednak można je stosować wyłącznie na małych etykietach, gdy nie ma miejsca na zamieszczenie pełnej nazwy pisanej czcionką wielkości minimum 7 punktów.
Zawartość produktu leczniczego w opakowaniu określa się, podając odpowiednio masę, objętość, liczbę dawek (liczbę dawek roztworu, liczbę rozpyleń inhalatora itp.), liczbę jednostek dawkowania lub wielkość opakowania.
4. Sposób podawania i droga podania
Określając drogę podania, należy korzystać z aktualnego ujednoliconego nazewnictwa Farmakopei Polskiej oraz z wykazu terminów standardowych Farmakopei Europejskiej. Niektóre z tych informacji są szczególnie potrzebne w przypadku produktów leczniczych dostępnych bez recepty.
5. Termin ważności
A. Określenie terminu ważności
Termin ważności należy wyrażać w sposób zrozumiały, niezaszyfrowany, z podaniem miesiąca i roku, przy czym miesiąc przedstawia się jako 2 cyfry lub co najmniej 3 litery, a rok – jako 4 cyfry. Termin ważności zamieszczony na produktach leczniczych, podający miesiąc i rok, należy rozumieć jako określenie ostatniego dnia danego miesiąca w tym roku.
B. Określenie okresu trwałości po otwarciu opakowania
Jeżeli trwałość danego produktu leczniczego jest zmniejszona, np. w wyniku rozcieńczania, zmieszania z rozpuszczalnikiem lub otwarcia opakowania, podaje się maksymalny okres ważności produktu leczniczego. Ponadto, jeżeli maksymalny okres przydatności zależy od sposobu rozpuszczania lub rodzaju rozpuszczalnika, w informacji na opakowaniu zamieszcza się zalecenie zapoznania się z informacją znajdującą się w ulotce.
W przypadku niektórych produktów, takich jak produkty radiofarmaceutyczne lub niektóre szczepionki, może być konieczne dokładniejsze określenie zarówno daty ważności, jak i czasu przechowywania po otwarciu pojemnika lub pierwszym użyciu produktu (podanie godziny, dnia, miesiąca i roku).
Na opakowaniach bezpośrednich zamiast określenia Termin ważności można użyć skrótu EXP z koniecznością objaśnienia znaczenia skrótu na opakowaniu zewnętrznym lub jeżeli to niemożliwe – w ulotce dla pacjenta w pkt 5: Termin ważności (EXP).
6. Warunki przechowywania
Warunki przechowywania w informacji na opakowaniu podaje się zgodnie z Charakterystyką Produktu Leczniczego. Jeżeli to konieczne, zamieszcza się dalsze krótkie wyjaśnienie dotyczące warunków przechowywania, w języku zrozumiałym dla pacjenta. Jeżeli jest to konieczne, zamieszcza się również ostrzeżenie o widocznych oznakach rozkładu produktu
7. Nazwa i adres podmiotu odpowiedzialnego; dotyczy podmiotu posiadającego pozwolenie na dopuszczenie do obrotu produktu leczniczego.
8. Numer (lub numery) pozwolenia; dotyczy numeru (numerów) pozwolenia na dopuszczenie do obrotu produktu leczniczego.
9. Numer serii; dotyczy numeru zwalnianej serii u wytwórcy.
Na opakowaniach bezpośrednich zamiast określenia Numer serii można użyć skrótu Lot z koniecznością objaśnienia znaczenia skrótu na opakowaniu zewnętrznym lub jeżeli to niemożliwe – w ulotce dla pacjenta w pkt 5: Numer serii (Lot).
10. Przykłady symboli lub piktogramów, które mogą być zamieszczane na opakowaniach:
1) piktogram znaku drogowego zakazującego 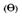
– produkty lecznicze silnie upośledzające sprawność psychofizyczną; bezwzględny zakaz prowadzenia pojazdów i obsługiwania urządzeń mechanicznych w ruchu przez 24 godziny po zastosowaniu;
2) piktogram znaku drogowego ostrzegawczego 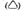
– produkty lecznicze, które mogą upośledzać sprawność psychofizyczną; jeżeli przepisana dawka i droga podania wskazują, że w okresie stosowania może pojawić się wyraźne upośledzenie sprawności psychofizycznej, to należy udzielić pacjentowi wskazówek co do konieczności zachowania szczególnej ostrożności podczas prowadzenia pojazdów lub obsługiwania urządzeń mechanicznych w ruchu lub o konieczności czasowego zaniechania takich czynności;
3) piktogram oka 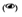
– produkty lecznicze podawane do oka;
4) piktogram ucha 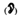
– produkty lecznicze podawane do ucha;
5) piktogram nosa – produkty lecznicze podawane do nosa;
6) piktogram profilu twarzy wraz z rozpylanym aerozolem – aerozole do wdychania;
7) piktogram radioaktywności – produkty lecznicze zawierające radionuklidy.
11. Do oznakowania opakowania produktu leczniczego stosuje się odpowiednio przepisy części II–III załącznika nr 2 do rozporządzenia, z zastrzeżeniem ust. 12. Informacje zamieszczane na oznakowaniu opakowań leków gotowych drukuje się czcionką wielkości co najmniej 7 punktów Didota lub taką, aby wysokość najmniejszej czcionki wynosiła 1,4 mm, z odstępem między wierszami wynoszącym co najmniej 3 mm.
12. Jeżeli w przypadku niektórych produktów leczniczych spełnienie wymagań wymienionych w części I załącznika nr 2 do rozporządzenia jest utrudnione, należy podejmować wszelkie możliwe działania w celu zwiększenia powierzchni oznakowania bez zmiany rozmiaru opakowania, np. etykietę można wydłużyć i poszerzyć albo tekst obrócić o 90° bez zmiany wielkości opakowania. Gdy wielkość etykiety jest z konieczności bardzo ograniczona, pierwszeństwo mają informacje dotyczące bezpieczeństwa stosowania produktu.
Załącznik nr 2
WYMAGANIA DOTYCZĄCE SPOSOBU SPORZĄDZANIA ULOTKI DLA PACJENTA
I. Rozmiar i rodzaj czcionki
Informacje zamieszczane w ulotkach leków gotowych drukuje się czcionką wielkości co najmniej 8 punktów Didota, z odstępem między wierszami wynoszącym co najmniej 3 mm.
Należy unikać pisania słów wielkimi literami (wersalikami). Rodzaj druku musi być łatwo czytelny.
II. Kolor druku
Litery należy drukować w takim kolorze, który wyraźnie odróżnia się od tła. Nagłówki można wyróżniać, drukując je w kolorze innym niż pozostały tekst. W kolorze czerwonym można drukować jedynie bardzo ważne ostrzeżenia.
III. Papier
Do długich ulotek sugeruje się wykorzystanie formatu A4 lub A5.
Nie należy stosować papieru o gramaturze mniejszej niż 40 g/m2.
IV. Zawartość ulotki
1. W ulotce dla pacjenta należy zamieszczać informacje zgodne z Charakterystyką Produktu Leczniczego, sformułowane w sposób łatwo zrozumiały dla pacjenta. Jeżeli używa się terminów naukowych lub specjalistycznych, podaje się ich wyjaśnienie. Należy posługiwać się terminologią z aktualnego ujednoliconego nazewnictwa Farmakopei Polskiej oraz z aktualnej Listy Terminologii Standardowej Farmakopei Europejskiej. Niekiedy jednak może być konieczne wyjaśnianie niektórych terminów w sposób zrozumiały dla pacjenta.
2. Nagłówki
Nagłówki i podtytuły należy wyróżnić. Można uniknąć powtarzania informacji poprzez wprowadzenie odnośników do innych części ulotki. Nagłówki należy wówczas ponumerować, aby w razie konieczności wprowadzenia odnośników do innych części ulotki łatwe było odnalezienie miejsca, w którym zawarta jest dana informacja. Stosowanie więcej niż dwóch poziomów podtytułów może wpłynąć niekorzystnie na czytelność ulotki.
3. Styl
Należy używać strony czynnej, formy bezosobowej. Jeżeli jest to tylko możliwe, należy unikać długich zdań (np. dłuższych niż 20 słów). Zaleca się, aby w wierszu było nie więcej niż 70 liter. Należy unikać zdań podrzędnie złożonych. Jeżeli podaje się wyliczenia, należy wyraźnie oddzielać poszczególne punkty; można używać kropek, przecinków, kresek, a także znaków specjalnych, jak duża czarna kropka. Grupę wyróżnianych pozycji zamieszcza się po dwukropku i kończy kropką. W wyliczeniach należy używać jak najmniejszej liczby słów i nie więcej niż jednego zdania. Na liście wyliczeń nie należy zamieszczać więcej niż 9 elementów pojedynczych lub nie więcej niż 5 elementów złożonych. Należy unikać skrótów. Nazwę produktu leczniczego można zastępować zaimkiem (np. on) dopóty, dopóki z kontekstu zdania jasno wynika, do czego się odnosi.
4. Wszędzie, gdzie jest to możliwe, należy wyjaśniać powody poszczególnych zaleceń. Zalecenie jednak należy podać na początku, np. „pacjenci z astmą powinni stosować produkt leczniczy X ostrożnie, gdyż może on spowodować napad duszności".
5. Dodatkowo można zamieszczać symbole lub piktogramy, określone w załączniku nr 1 do rozporządzenia, ale tylko, gdy dzięki nim informacja jest bardziej zrozumiała dla pacjenta. Piktogramy nie mogą zawierać elementów reklamy.
6. Należy unikać nieuzasadnionego używania wersalików (wielkich liter), ponieważ zmniejsza to czytelność pozostałych informacji. Wersaliki można jednak stosować w celu podkreślenia ważnej informacji.
V. Grupy produktów
Jeżeli produkty różnią się zawartością substancji czynnej i postacią farmaceutyczną, należy opracować do nich osobne ulotki. W niektórych przypadkach może być jednak celowe zamieszczanie w ulotce informacji o innych dostępnych postaciach i dawkach tego samego produktu leczniczego, jeżeli identyczne są:
1) wskazania;
2) dawkowanie;
3) droga podania;
4) przeciwwskazania, działania niepożądane, ostrzeżenia i środki ostrożności.
Również w przypadku produktów leczniczych dostępnych bez recepty może być celowe odniesienie do innych postaci farmaceutycznych, np. w ulotce do tabletek, które nie są zalecane dla dzieci, można wskazać, że istnieje również roztwór do podawania doustnego.
VI. Produkty nieprzeznaczone do samodzielnego stosowania przez pacjentów
1. Do produktów leczniczych podawanych w szpitalu można dołączyć osobne ulotki do rozprowadzania wśród pacjentów. Mogą mieć one na przykład formę bloczka ulotek z oddzielnymi kartkami do wyrywania, dołączonego do opakowania produktu leczniczego razem z Charakterystyką Produktu Leczniczego przeznaczoną dla personelu szpitalnego. Jeżeli ulotka dla pacjenta jest dostarczana osobno, wytwórca produktu leczniczego powinien przedsięwziąć odpowiednie kroki w celu umożliwienia personelowi szpitalnemu dostarczania pacjentom aktualnej wersji ulotki.
2. W przypadku produktów leczniczych podawanych przez personel medyczny informacje pochodzące z Charakterystyki Produktu Leczniczego przeznaczone dla personelu szpitalnego (w tym instrukcja dotycząca podania) mogą być zamieszczone na końcu ulotki w formie fragmentu kartki nadającej się do oderwania przed wręczeniem właściwej ulotki pacjentowi.
- Data ogłoszenia: 2009-03-13
- Data wejścia w życie: 2009-03-13
- Data obowiązywania: 2014-01-01
- Dokument traci ważność: 2015-08-04

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA