REKLAMA
Dziennik Ustaw - rok 2008 nr 201 poz. 1247
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 4 listopada 2008 r.
w sprawie wzorów dokumentów przedkładanych w związku z badaniem klinicznym produktu leczniczego oraz w sprawie wysokości i sposobu uiszczania opłat za rozpoczęcie badania klinicznego
Na podstawie art. 37w ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne (Dz. U. z 2008 r. Nr 45, poz. 271) zarządza się, co następuje:
1) wzór wniosku do komisji bioetycznej o wydanie opinii o badaniu klinicznym produktu leczniczego oraz do ministra właściwego do spraw zdrowia o rozpoczęcie badania klinicznego produktu leczniczego;
2) dokumentację stanowiącą podstawę wydania opinii o badaniu klinicznym produktu leczniczego przez komisję bioetyczną;
3) dokumentację, którą należy dołączyć do wniosku do ministra właściwego do spraw zdrowia o rozpoczęcie badania klinicznego produktu leczniczego;
4) wzór wniosku do komisji bioetycznej o wydanie opinii i do ministra właściwego do spraw zdrowia o wyrażenie zgody w zakresie istotnych i mających wpływ na bezpieczeństwo uczestników badania klinicznego zmian w protokole badania klinicznego lub dokumentacji dotyczącej badanego produktu leczniczego będącej podstawą uzyskania pozwolenia na prowadzenie badania klinicznego;
5) wzór zawiadomienia do komisji bioetycznej i do ministra właściwego do spraw zdrowia o zakończeniu badania klinicznego produktu leczniczego;
6) wysokość i sposób uiszczania opłat za rozpoczęcie badania klinicznego.
1) państwie członkowskim – rozumie się przez to terytorium państwa członkowskiego Unii Europejskiej wchodzące w skład Unii Europejskiej oraz terytorium państwa członkowskiego Europejskiego Porozumienia o Wolnym Handlu (EFTA) – strony umowy o Europejskim Obszarze Gospodarczym;
2) pacjencie – rozumie się również uczestnika badania klinicznego.
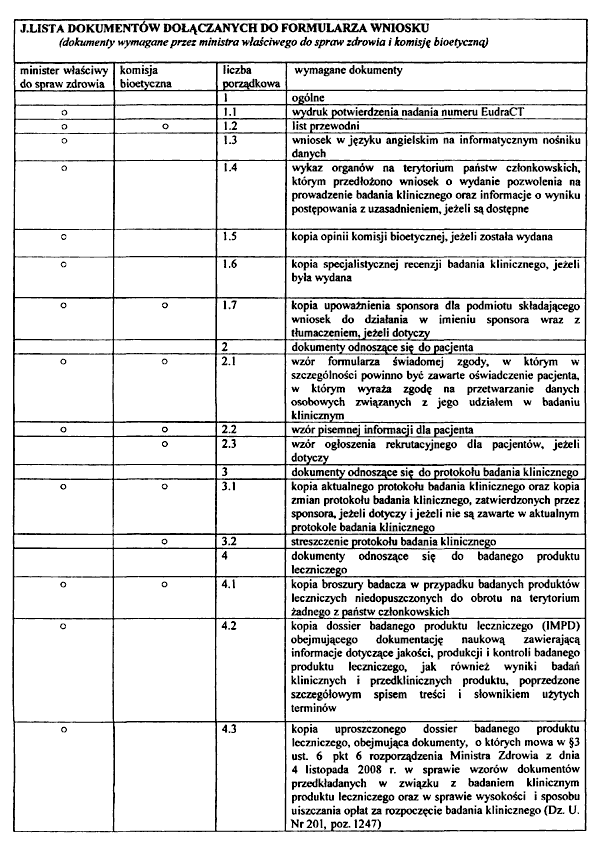
2. Do wniosku o wydanie opinii o badaniu klinicznym przez komisję bioetyczną dołącza się:
1) list przewodni;
2) kopię aktualnego protokołu badania klinicznego;
3) kopię zmian protokołu badania klinicznego, zatwierdzonych przez sponsora, jeżeli dotyczy i jeżeli nie są zawarte w aktualnym protokole badania klinicznego;
4) streszczenie protokołu badania klinicznego;
5) kopię broszury badacza w przypadku badanych produktów leczniczych niedopuszczonych do obrotu na terytorium żadnego z państw członkowskich;
6) kopię upoważnienia sponsora dla podmiotu składającego wniosek do działania w imieniu sponsora wraz z tłumaczeniem, jeżeli dotyczy;
7) kopię dokumentu upoważniającego organizację prowadzącą badanie kliniczne na zlecenie, określającego zakres uprawnień i obowiązków tego podmiotu wraz z tłumaczeniem, jeżeli dotyczy;
8) wzór formularza świadomej zgody, w którym w szczególności powinno być zawarte oświadczenie pacjenta, w którym wyraża zgodę na przetwarzanie danych osobowych związanych z jego udziałem w badaniu klinicznym;
9) wzór pisemnej informacji dla pacjenta;
10) wzór karty obserwacji klinicznej;
11) życiorys badacza, w tym opis jego działalności naukowej i zawodowej;
12) oświadczenie sponsora albo upoważnionego przez niego podmiotu, o którym mowa w pkt 7 lub ust. 6 pkt 25, albo badacza, dotyczące zasad rekrutacji pacjentów, o ile nie zostało zawarte w protokole badania klinicznego;
13) wzór ogłoszenia rekrutacyjnego dla pacjentów, jeżeli dotyczy;
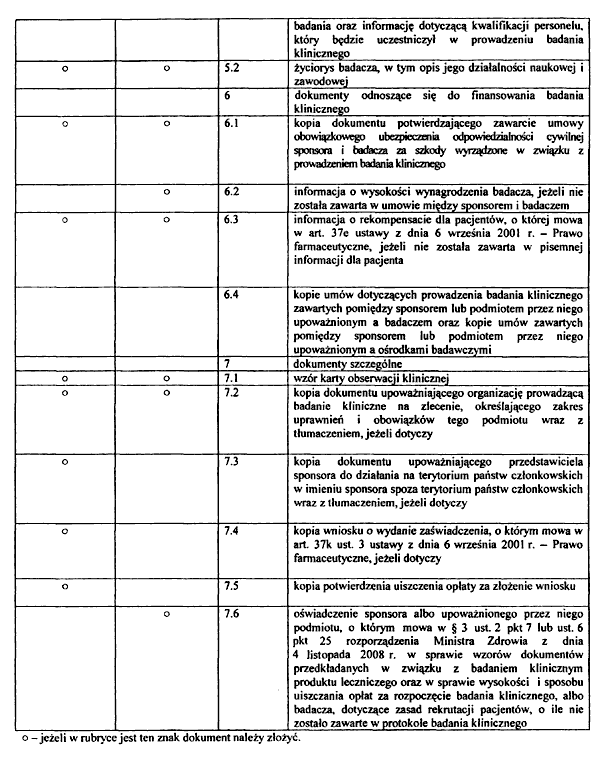
14) oświadczenie badacza dotyczące wyposażenia ośrodka badawczego w zakresie niezbędnym do przeprowadzenia badania oraz informację dotyczącą kwalifikacji personelu, który będzie uczestniczył w prowadzeniu badania klinicznego;
15) informację o rekompensacie dla pacjenta, o której mowa w art. 37e ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, jeżeli nie została zawarta w pisemnej informacji dla pacjenta;
16) kopię dokumentu potwierdzającego zawarcie umowy obowiązkowego ubezpieczenia odpowiedzialności cywilnej sponsora i badacza za szkody wyrządzone w związku z prowadzeniem badania klinicznego;
17) informację o wysokości wynagrodzenia badacza, jeżeli nie została zawarta w umowie między sponsorem i badaczem.
3. Do wniosku, o którym mowa w § 1 pkt 1, składanego do komisji bioetycznej dołącza się kopie umów dotyczących prowadzenia badania klinicznego zawartych pomiędzy sponsorem lub podmiotem przez niego upoważnionym a badaczem oraz kopie umów zawartych pomiędzy sponsorem lub podmiotem przez niego upoważnionym a ośrodkami badawczymi, potwierdzonych za zgodność z oryginałem przez sponsora albo podmiot przez niego upoważniony do działania w jego imieniu.
4. Kopie umów, o których mowa w ust. 3, dołącza się niezwłocznie po ich zawarciu, nie później niż przed dniem wydania przez komisję bioetyczną opinii o badaniu klinicznym.
5. Badania klinicznego nie można rozpocząć w ośrodku badawczym, jeżeli kopia umowy, o której mowa w ust. 3, zawarta z danym ośrodkiem nie została dołączona do dnia wydania opinii o badaniu klinicznym.
6. Do wniosku o rozpoczęcie badania klinicznego dołącza się:
1) wydruk potwierdzenia nadania numeru EudraCT;
2) list przewodni;
3) wniosek w języku angielskim na informatycznym nośniku danych, jako dokument elektroniczny w formacie xml do bazy EudraCT, zgodnie ze wzorem dostępnym na stronie internetowej Europejskiej Agencji Leków, przygotowanym na podstawie dyrektywy 2001/20/WE Parlamentu Europejskiego i Rady z dnia 4 kwietnia 2001 r. w sprawie zbliżenia przepisów ustawowych, wykonawczych i administracyjnych państw członkowskich, odnoszących się do wdrożenia zasad dobrej praktyki klinicznej w prowadzeniu badań klinicznych produktów leczniczych, przeznaczonych do stosowania u ludzi (Dz. Urz. WE L 121 z 01.05.2001, str. 34; Dz. Urz. UE Polskie wydanie specjalne, rozdz. 13, t. 26, str. 299);
4) kopię broszury badacza w przypadku badanych produktów leczniczych niedopuszczonych do obrotu na terytorium żadnego z państw członkowskich;
5) kopię dossier badanego produktu leczniczego (IMPD) obejmującego dokumentację naukową zawierającą informacje dotyczące jakości, produkcji i kontroli badanego produktu leczniczego, jak również wyniki badań klinicznych i przedklinicznych produktu, poprzedzone szczegółowym spisem treści i słownikiem użytych terminów;
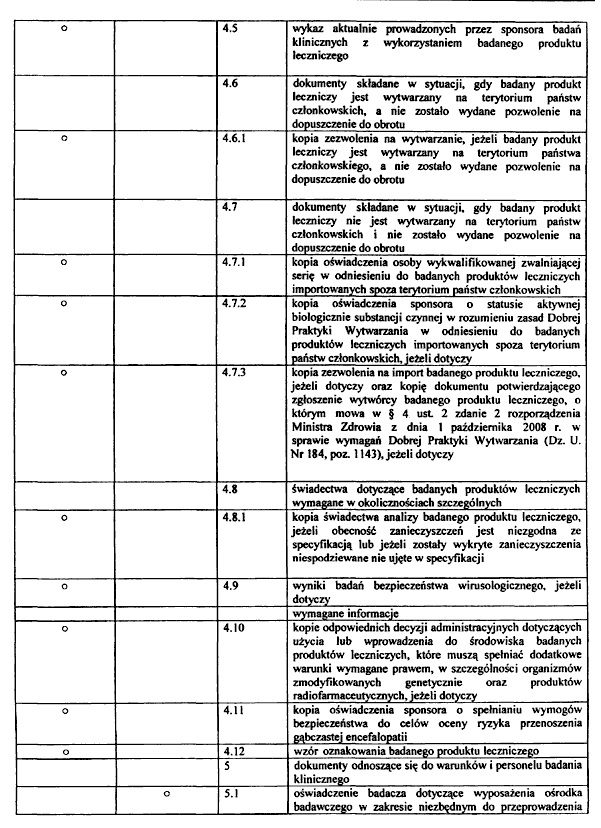
6) kopię uproszczonego dossier badanego produktu leczniczego:
a) obejmującego kopię Charakterystyki Produktu Leczniczego – w przypadku badanych produktów leczniczych dopuszczonych do obrotu na terytorium przynajmniej jednego z państw członkowskich i stosowanych w badaniu klinicznym zgodnie z Charakterystyką Produktu Leczniczego, a w przypadku gdy badany produkt leczniczy stosowany jest poza wskazaniem zawartym w Charakterystyce Produktu Leczniczego – dodatkowo wyniki badań klinicznych i przedklinicznych przeprowadzonych z użyciem badanego produktu leczniczego,
b) obejmującego wyniki badań klinicznych i przedklinicznych oraz informacje dotyczące jakości badanego produktu leczniczego – w przypadku badanych produktów leczniczych dopuszczonych jako produkt leczniczy do obrotu na terytorium przynajmniej jednego z państw członkowskich w innej postaci lub mocy,
c) obejmującego wyniki badań klinicznych i przedklinicznych oraz informacje dotyczące jakości badanego produktu leczniczego – w przypadku badanych produktów leczniczych niedopuszczonych do obrotu na terytorium żadnego z państw członkowskich, a których wszystkie substancje czynne wchodzą w skład produktów leczniczych dopuszczonych do obrotu na terytorium przynajmniej jednego z państw członkowskich; jeżeli substancje czynne pochodzą od innego wytwórcy, konieczne jest dołączenie danych na ich temat,
d) w przypadku badanego produktu leczniczego objętego innym wnioskiem o rozpoczęcie badania klinicznego, obejmującego nowe wyniki badań klinicznych i przedklinicznych oraz informacje dotyczące jakości badanego produktu leczniczego uzyskane od czasu złożenia poprzedniego wniosku lub tylko informację o numerze EudraCT badania objętego wspomnianym wnioskiem, jeśli takich wyników i informacji nie uzyskano,
e) obejmującego informacje dotyczące jakości badanego produktu leczniczego – w przypadku placebo;
7) kopię upoważnienia sponsora dla podmiotu składającego wniosek do działania w imieniu sponsora wraz z tłumaczeniem, jeżeli dotyczy;
8) wzór karty obserwacji klinicznej;
9) wzór formularza świadomej zgody, w którym w szczególności powinno być zawarte oświadczenie pacjenta, w którym wyraża zgodę na przetwarzanie danych osobowych związanych z jego udziałem w badaniu klinicznym;
10) wzór pisemnej informacji dla pacjenta;
11) kopię aktualnego protokołu badania klinicznego;
12) kopię zmian protokołu badania klinicznego, zatwierdzonych przez sponsora, jeżeli dotyczy i jeżeli nie są zawarte w aktualnym protokole badania klinicznego;
13) kopię specjalistycznej recenzji badania klinicznego, jeżeli była wydana;
14) kopię świadectwa analizy badanego produktu leczniczego, jeżeli obecność zanieczyszczeń jest niezgodna ze specyfikacją lub jeżeli zostały wykryte zanieczyszczenia niespodziewane nieujęte w specyfikacji;
15) kopię zezwolenia na wytwarzanie, jeżeli badany produkt leczniczy jest wytwarzany na terytorium państwa członkowskiego, a nie zostało wydane pozwolenie na dopuszczenie do obrotu;
16) kopię zezwolenia na import badanego produktu leczniczego, jeżeli dotyczy, oraz kopię dokumentu potwierdzającego zgłoszenie wytwórcy badanego produktu leczniczego, o którym mowa w § 4 ust. 2 zdanie drugie rozporządzenia Ministra Zdrowia z dnia 1 października 2008 r. w sprawie wymagań Dobrej Praktyki Wytwarzania (Dz. U. Nr 184, poz. 1143), jeżeli dotyczy;
17) kopię oświadczenia osoby wykwalifikowanej zwalniającej serię w odniesieniu do badanych produktów leczniczych importowanych spoza terytorium państw członkowskich;
18) kopię oświadczenia sponsora o statusie aktywnej biologicznie substancji czynnej w rozumieniu zasad Dobrej Praktyki Wytwarzania w odniesieniu do badanych produktów leczniczych importowanych spoza terytorium państw członkowskich, jeżeli dotyczy;
19) kopie odpowiednich decyzji administracyjnych dotyczących użycia lub wprowadzenia do środowiska badanych produktów leczniczych, które muszą spełniać dodatkowe warunki wymagane prawem, w szczególności organizmów zmodyfikowanych genetycznie oraz produktów radiofarmaceutycznych, jeżeli dotyczy;
20) wyniki badań bezpieczeństwa wirusologicznego, jeżeli dotyczy;
21) kopię oświadczenia sponsora o spełnianiu wymogów bezpieczeństwa do celów oceny ryzyka przenoszenia gąbczastej encefalopatii, jeżeli dotyczy;
22) życiorys badacza, w tym opis jego działalności naukowej i zawodowej;
23) kopię dokumentu potwierdzającego zawarcie umowy obowiązkowego ubezpieczenia odpowiedzialności cywilnej sponsora i badacza za szkody wyrządzone w związku z prowadzeniem badania klinicznego;
24) informację o rekompensacie dla pacjentów, o której mowa w art. 37e ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, jeżeli nie została zawarta w informacji dla pacjenta;
25) kopię dokumentu upoważniającego organizację prowadzącą badanie kliniczne na zlecenie, określającego zakres uprawnień i obowiązków tego podmiotu wraz z tłumaczeniem, jeżeli dotyczy;
26) kopię dokumentu upoważniającego przedstawiciela sponsora do działania na terytorium państw członkowskich w imieniu sponsora spoza terytorium państw członkowskich wraz z tłumaczeniem, jeżeli dotyczy;
27) wykaz aktualnie prowadzonych przez sponsora badań klinicznych z wykorzystaniem badanego produktu leczniczego;
28) wykaz organów na terytorium państw członkowskich, którym przedłożono wniosek o wydanie pozwolenia na prowadzenie badania klinicznego oraz informacje o wyniku postępowania z uzasadnieniem, jeżeli są dostępne;
29) wzór oznakowania badanego produktu leczniczego;
30) kopię wniosku o wydanie zaświadczenia, o którym mowa w art. 37k ust. 3 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, jeżeli dotyczy;
31) kopię opinii komisji bioetycznej, jeżeli została wydana;
32) kopię potwierdzenia uiszczenia opłaty za złożenie wniosku.
7. Do wniosku, o którym mowa w § 1 pkt 1, składanego do ministra właściwego do spraw zdrowia, dołącza się kopie umów dotyczących prowadzenia badania klinicznego zawartych pomiędzy sponsorem lub podmiotem przez niego upoważnionym a badaczem oraz kopie umów zawartych pomiędzy sponsorem lub podmiotem przez niego upoważnionym a ośrodkami badawczymi, potwierdzonych za zgodność z oryginałem przez sponsora albo podmiot przez niego upoważniony do działania w jego imieniu.
8. Kopie umów, o których mowa w ust. 7, dołącza się niezwłocznie po ich zawarciu, nie później niż przed wydaniem przez ministra właściwego do spraw zdrowia pozwolenia na prowadzenie badania klinicznego lub przed upływem terminu, o którym mowa w art. 37p ust. 1 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne.
9. Dołączenie kopii umów w terminie, o którym mowa w ust. 8, pozostaje bez wpływu na rozpoczęcie biegu terminu wskazanego w art. 37p ust. 1 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne.
10. Badania klinicznego nie można rozpocząć w ośrodku badawczym, jeżeli kopia umowy, o której mowa w ust. 7, zawarta z danym ośrodkiem nie została dołączona w terminie, o którym mowa w ust. 8.
11. Kopie dokumentów wymienione w § 3 powinny zostać sporządzone w formie papierowej, z zastrzeżeniem ust. 6 pkt 3.
2. Do wniosku, o którym mowa w ust. 1, składanym do ministra właściwego do spraw zdrowia dołącza się wniosek w języku angielskim na informatycznym nośniku danych, jako dokument elektroniczny w formacie doc do bazy EudraCT, zgodnie ze wzorem dostępnym na stronie internetowej Europejskiej Agencji Leków przygotowanym na podstawie dyrektywy 2001/20/WE Parlamentu Europejskiego i Rady z dnia 4 kwietnia 2001 r. w sprawie zbliżenia przepisów ustawowych, wykonawczych i administracyjnych państw członkowskich, odnoszących się do wdrożenia zasad dobrej praktyki klinicznej w prowadzeniu badań klinicznych produktów leczniczych, przeznaczonych do stosowania u ludzi.
2. Do zawiadomienia, o którym mowa w ust. 1, dołącza się zawiadomienie w języku angielskim na informatycznym nośniku danych, jako dokument elektroniczny w formacie doc do bazy EudraCT, zgodnie ze wzorem dostępnym na stronie internetowej Europejskiej Agencji Leków przygotowanym na podstawie dyrektywy 2001/20/WE Parlamentu Europejskiego i Rady z dnia 4 kwietnia 2001 r. w sprawie zbliżenia przepisów ustawowych, wykonawczych i administracyjnych państw członkowskich, odnoszących się do wdrożenia zasad dobrej praktyki klinicznej w prowadzeniu badań klinicznych produktów leczniczych, przeznaczonych do stosowania u ludzi.
2. Dokumenty, o których mowa w § 3 ust. 2 pkt 1, 4, 8, 9, 13–17 oraz w ust. 6 pkt 2, 9, 10, 14, 23, 24, 29, dołącza się w języku polskim.
3. Dokumenty inne niż wymienione w ust. 1 i 2 składa się w języku polskim lub angielskim.
2. Opłaty za złożenie wniosku, o którym mowa w ust. 1, uiszcza się na rachunek Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych gotówką, przelewem lub przekazem pocztowym.
3. Opłaty należne z różnych tytułów uiszcza się oddzielnie.
Minister Zdrowia: E. Kopacz
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 16 listopada 2007 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 216, poz. 1607).
2) Niniejsze rozporządzenie było poprzedzone rozporządzeniem Ministra Zdrowia z dnia 3 stycznia 2007 r. w sprawie wzoru wniosku o rozpoczęcie badania klinicznego produktu leczniczego oraz o wydanie przez komisję bioetyczną opinii o badaniu klinicznym produktu leczniczego (Dz. U. Nr 6, poz. 46) oraz rozporządzeniem Ministra Zdrowia z dnia 3 marca 2006 r. w sprawie wysokości i sposobu uiszczania opłat za rozpoczęcie badania klinicznego (Dz. U. Nr 45, poz. 321), które utraciły moc z dniem 1 listopada 2008 r. na podstawie art. 15 ustawy z dnia 30 marca 2007 r. o zmianie ustawy – Prawo farmaceutyczne oraz o zmianie niektórych innych ustaw (Dz. U. Nr 75, poz. 492).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 4 listopada 2008 r. (poz. 1247)
Załącznik nr 1

















Załącznik nr 2






Załącznik nr 3


Załącznik nr 4

- Data ogłoszenia: 2008-11-13
- Data wejścia w życie: 2008-11-13
- Data obowiązywania: 2008-11-13
- Dokument traci ważność: 2012-05-02

REKLAMA