REKLAMA
Dziennik Ustaw - rok 2008 nr 189 poz. 1157
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 15 października 2008 r.
w sprawie sposobu przeprowadzania kontroli i inspekcji przez Państwową Inspekcję Farmaceutyczną
Na podstawie art. 123 ust. 2 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne (Dz. U. z 2008 r. Nr 45, poz. 271) zarządza się, co następuje:
1) sposób przeprowadzania kontroli i inspekcji przez Państwową Inspekcję Farmaceutyczną;
2) rodzaje kontroli i inspekcji;
3) sposób i tryb pobierania próbek do badań oraz przeprowadzania badań;
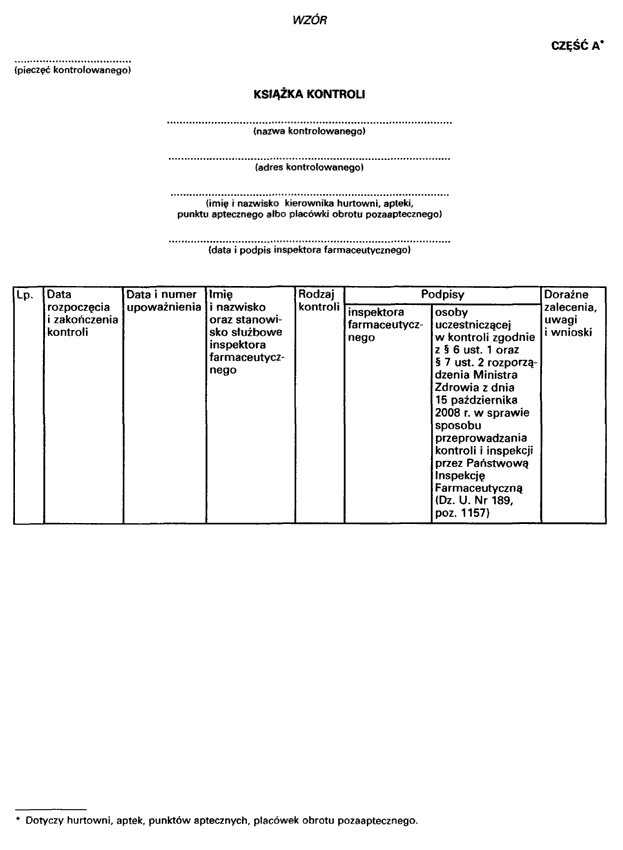
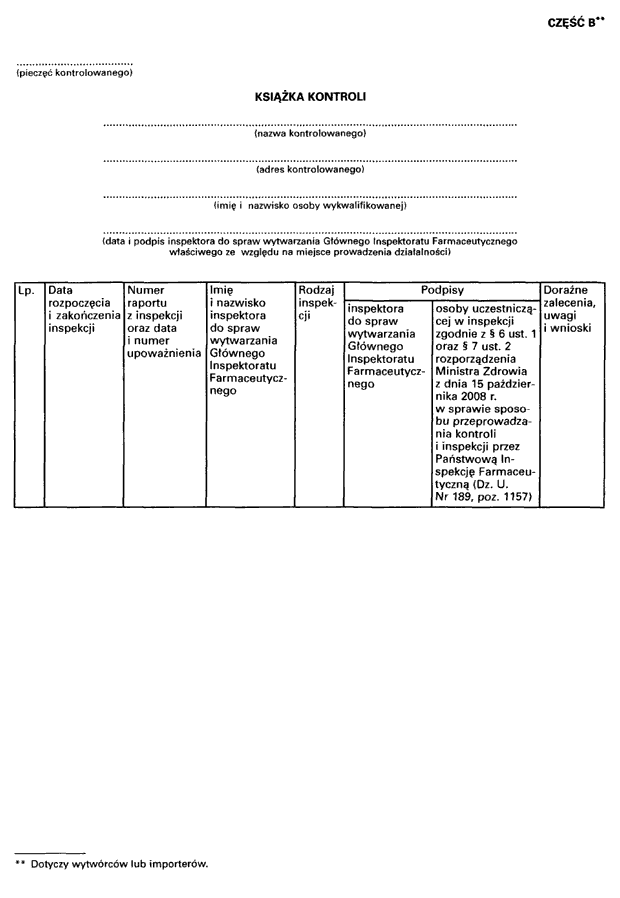
4) wzór i sposób prowadzenia książki kontroli, sposób dokonywania wpisów oraz tryb powiadamiania o usunięciu stwierdzonych uchybień;
5) tryb przeprowadzania kontroli przyjmowanych i wydawanych produktów leczniczych i wyrobów medycznych oraz warunki ich transportowania.
1) ustawie – rozumie się przez to ustawę z dnia 6 września 2001 r. – Prawo farmaceutyczne;
2) inspektorze – rozumie się przez to inspektora farmaceutycznego lub inspektora do spraw wytwarzania Głównego Inspektoratu Farmaceutycznego;
3) kontrolowanym – rozumie się przez to podmiot podlegający kontroli albo inspekcji zgodnie z przepisami ustawy.
1) planowe – umieszczone w planie kontroli albo inspekcji, przeprowadzane w celu potwierdzenia spełniania obowiązków wynikających z ustawy;
2) sprawdzające – przeprowadzane w celu skontrolowania wykonania działań naprawczych;
3) doraźne – przeprowadzane w przypadku powzięcia uzasadnionego podejrzenia o uchybieniach kontrolowanego, powodujących zagrożenie dla jakości, bezpieczeństwa stosowania lub skuteczności produktów leczniczych i wyrobów medycznych.
2. Kontrolę przeprowadza inspektor farmaceutyczny na podstawie upoważnienia wydanego przez wojewódzkiego inspektora farmaceutycznego.
2. Jeden egzemplarz upoważnienia pozostawia się kontrolowanemu.
3. Rozpoczęcie kontroli albo inspekcji inspektor potwierdza, dokonując odpowiedniego wpisu w książce kontroli, o której mowa w art. 123 ust. 1 ustawy.
4. Inspektor do spraw wytwarzania Głównego Inspektoratu Farmaceutycznego, dokonując inspekcji planowej wytwórni produktów leczniczych lub miejsca prowadzenia działalności importowej, przed rozpoczęciem inspekcji podaje kontrolowanemu plan przebiegu inspekcji, w tym przewidywany okres prowadzenia inspekcji, zakres inspekcji oraz wykaz personelu zobowiązanego do składania wyjaśnień istotnych dla prowadzonej inspekcji.
2. Oświadczenie o rezygnacji z prawa uczestniczenia w czynnościach podejmowanych w ramach kontroli albo inspekcji jest składane na piśmie. W razie odmowy złożenia oświadczenia inspektor dokonuje odpowiedniej adnotacji w protokole z kontroli albo w raporcie z inspekcji.
2. W kontroli albo inspekcji mogą brać udział biegli lub eksperci na podstawie imiennego upoważnienia wydanego odpowiednio przez wojewódzkiego inspektora farmaceutycznego albo Głównego Inspektora Farmaceutycznego.
2. Protokół z kontroli albo raport z inspekcji zawierają w szczególności:
1) wskazanie kontrolowanego;
2) wskazanie inspektorów;
3) wskazanie biegłych i ekspertów, o których mowa w § 7 ust. 2 – jeżeli brali udział w kontroli albo inspekcji;
4) określenie przedmiotu i zakresu kontroli albo inspekcji;
5) określenie miejsca i czasu kontroli albo inspekcji;
6) opis dokonanych ustaleń;
7) przedstawienie dowodów;
8) pouczenie o prawie złożenia zastrzeżeń lub wyjaśnień, o których mowa w § 9 ust. 2 i § 10 ust. 2.
3. Protokół z kontroli podpisują:
1) kontrolowany albo osoba przez niego upoważniona;
2) inspektorzy przeprowadzający kontrolę;
3) osoby, których wyjaśnienia jako istotne dla czynności kontrolnych przytoczone zostały w protokole.
4. Raport z inspekcji podpisuje inspektor do spraw wytwarzania Głównego Inspektoratu Farmaceutycznego lub inspektorzy do spraw wytwarzania Głównego Inspektoratu Farmaceutycznego przeprowadzający inspekcję.
2. Kontrolowany, który nie zgadza się z ustaleniami protokołu z kontroli, może w terminie 3 dni od dnia jego doręczenia złożyć pisemnie zastrzeżenia lub wyjaśnienia, wskazując jednocześnie stosowne wnioski dowodowe. Inspektor jest obowiązany rozpatrzyć zgłoszone zastrzeżenia w terminie 7 dni od dnia ich otrzymania. W przypadku uwzględnienia zastrzeżeń inspektor uzupełnia protokół z kontroli i przedstawia go ponownie do podpisu kontrolowanego albo osoby przez niego upoważnionej lub osób, o których mowa w § 8 ust. 3 pkt 3.
3. W przypadku odmowy podpisania protokołu z kontroli odmawiający składa pisemne wyjaśnienie co do przyczyn odmowy.
4. O odmowie podpisania protokołu z kontroli, przyczynie tej odmowy oraz o złożeniu wyjaśnień inspektor dokonuje wzmianki w protokole z kontroli.
2. Kontrolowany, który nie zgadza się z ustaleniami raportu z inspekcji, może w terminie 3 dni od dnia jego doręczenia złożyć zastrzeżenia lub wyjaśnienia, wskazując jednocześnie stosowne wnioski dowodowe. Inspektor jest obowiązany rozpatrzyć zgłoszone zastrzeżenia w terminie 7 dni od dnia ich otrzymania. W przypadku uwzględnienia zastrzeżeń inspektor uzupełnia raport z inspekcji i przekazuje go ponownie kontrolowanemu.
2. Wpis w książce kontroli nie może być wymazywany ani w inny sposób usuwany.
3. Inspektor może dokonywać skreśleń i poprawek we wpisie w taki sposób, aby wyrazy skreślone i poprawione były czytelne.
4. Skreślenia i poprawki powinny być dokonane przed podpisaniem wpisu.
5. O dokonaniu skreśleń i poprawek należy na końcu wpisu sporządzić adnotację z określeniem strony książki oraz ich treści.
2. O usunięciu stwierdzonych uchybień wynikających z inspekcji kontrolowany pisemnie informuje Głównego Inspektora Farmaceutycznego.
2. Próbki do badań pobiera się w ilości niezbędnej do przeprowadzenia właściwego badania laboratoryjnego.
3. Protokół pobrania próbek sporządza się w trzech egzemplarzach, z których jeden pozostawia się kontrolowanemu, drugi dołącza się do próbki kierowanej do badań jakościowych, trzeci pozostawia się w dokumentacji inspektora.
2. Do próbki przesyłanej do badań należy dołączyć protokół, o którym mowa w § 13, oraz sporządzony przez inspektora wniosek o przeprowadzenie badań laboratoryjnych.
2. Przepis ust. 1 nie dotyczy leku recepturowego.
3. Próbka pozostawiona na przechowanie zgodnie z ust. 1 powinna być przechowywana u kontrolowanego do dnia otrzymania orzeczenia, o którym mowa w § 16.
2. Po przeprowadzeniu badań jakościowych jednostka prowadząca badania laboratoryjne sporządza orzeczenie o wyniku badań jakościowych próbek pobranych przez inspektora podczas kontroli albo inspekcji.
3. Jednostka prowadząca badania laboratoryjne przekazuje orzeczenie, o którym mowa w ust. 2, inspektorowi, który wystąpił z wnioskiem o przeprowadzenie badania, oraz kontrolowanemu.
2. Temperatura w przestrzeniach transportowych powinna odpowiadać wymaganiom określonym przez podmiot odpowiedzialny lub autoryzowanego przedstawiciela w dokumentacji produktu lub wyrobu.
Minister Zdrowia: E. Kopacz
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 16 listopada 2007 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 216, poz. 1607).
Załącznik do rozporządzenia Ministra Zdrowia
z dnia 15 października 2008 r. (poz. 1157)
- Data ogłoszenia: 2008-10-23
- Data wejścia w życie: 2008-10-31
- Data obowiązywania: 2008-10-31
- Dokument traci ważność: 2009-08-29

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA