REKLAMA
Dziennik Ustaw - rok 2004 nr 37 poz. 334
ROZPORZĄDZENIE MINISTRA ROLNICTWA l ROZWOJU WSI1)
z dnia 13 lutego 2004 r.
w sprawie metod analiz niektórych produktów i półproduktów przemysłu cukrowniczego
Na podstawie art. 34 pkt 2 ustawy z dnia 21 grudnia 2000 r. o jakości handlowej artykułów rolno-spożywczych (Dz. U. z 2001 r. Nr 5, poz. 44, z późn. zm.2)) zarządza się, co następuje:
1) cukru (cukru białego) i cukru ekstrabiałego (cukru rafinowanego) – w zakresie polaryzacji, zawartości cukru inwertowanego i wilgotności;
2) cukru przemysłowego – w zakresie wilgotności, polaryzacji, zawartości cukru inwertowanego;
3) płynnego cukru (roztworu cukru), płynnego cukru inwertowanego (roztworu cukru inwertowanego) oraz syropu cukru inwertowanego – w zakresie zawartości suchej masy i zawartości cukrów redukujących;
4) syropu glukozowego oraz syropu glukozowego w proszku – w zakresie zawartości suchej masy, równoważnika dekstrozy i zawartości popiołu siarczanowego;
5) jednowodnej glukozy (jednowodnej dekstrozy) i bezwodnej glukozy (bezwodnej dekstrozy) – w zakresie zawartości dekstrozy (D-glukozy), suchej masy i popiołu siarczanowego;
6) fruktozy – w zakresie wilgotności.
2. Zawartość cukru inwertowanego w cukrze przemysłowym oznacza się zgodnie z metodą określoną w załączniku nr 2 do rozporządzenia.
3. Zawartość cukru inwertowanego w cukrze (cukrze białym) i w cukrze ekstrabiałym (cukrze rafinowanym) oznacza się zgodnie z metodą określoną w załączniku nr 3 do rozporządzenia.
4. Zawartość cukrów redukujących w płynnym cukrze (roztworze cukru), w płynnym cukrze inwertowanym (roztworze cukru inwertowanego) i w syropie cukru inwertowanego oznacza się zgodnie z metodą wybraną spośród metod określonych w załączniku nr 4 do rozporządzenia.
5. Równoważnik dekstrozy w syropie glukozowym i syropie glukozowym w proszku oraz zawartość dekstrozy (D-glukozy) w jednowodnej glukozie (jednowodnej dekstrozie) i w bezwodnej glukozie (bezwodnej dekstrozie) oznacza się zgodnie z metodą l określoną w załączniku nr 4 do rozporządzenia albo zgodnie z metodą określoną w załączniku nr 5 do rozporządzenia.
6. Wilgotność cukru (cukru białego), cukru ekstrabiałego (cukru rafinowanego), cukru przemysłowego i fruktozy oznacza się zgodnie z metodą określoną w załączniku nr 6 do rozporządzenia.
7. Zawartość suchej masy w płynnym cukrze (roztworze cukru), płynnym cukrze inwertowanym (roztworze cukru inwertowanego) i w syropie cukru inwertowanego oznacza się zgodnie z metodą określoną w załączniku nr 7 do rozporządzenia.
8. Zawartość suchej masy w syropie glukozowym, syropie glukozowym w proszku, w jednowodnej glukozie (jednowodnej dekstrozie) i w bezwodnej glukozie (bezwodnej dekstrozie) oznacza się zgodnie z metodą określoną w załączniku nr 8 do rozporządzenia.
9. Zawartość popiołu siarczanowego w syropie glukozowym, syropie glukozowym w proszku, w jednowodnej glukozie (jednowodnej dekstrozie) i w bezwodnej glukozie (bezwodnej dekstrozie) oznacza się zgodnie z metodą określoną w załączniku nr 9 do rozporządzenia.
Minister Rolnictwa i Rozwoju Wsi: W. Olejniczak
|
|
1) Minister Rolnictwa i Rozwoju Wsi kieruje działem administracji rządowej – rynki rolne, na podstawie § 1 ust. 2 pkt 3 rozporządzenia Prezesa Rady Ministrów z dnia 29 marca 2002 r. w sprawie szczegółowego zakresu działania Ministra Rolnictwa i Rozwoju Wsi (Dz. U. Nr 32, poz. 305).
2) Zmiany wymienionej ustawy zostały ogłoszone w Dz. U. z 2001 r. Nr 154, poz. 1802, z 2002 r. Nr 135, poz. 1145 i Nr 166, poz. 1360 oraz z 2003 r. Nr 208, poz. 2020 i Nr 223, poz. 2220 i 2221.
Załączniki do rozporządzenia Ministra Rolnictwa i Rozwoju Wsi
z dnia 13 lutego 2004 r. (poz. 334)
Załącznik nr 1
METODA OZNACZANIA POLARYZACJI W CUKRZE (CUKRZE BIAŁYM), W CUKRZE EKSTRABIAŁYM (CUKRZE RAFINOWANYM) ORAZ W CUKRZE PRZEMYSŁOWYM
1. Polaryzacja polega na skręceniu płaszczyzny światła spolaryzowanego przechodzącego przez roztwór badanego cukru. Przyjmuje się, że polaryzacja wynosi w stopniach sacharymetrycznych 100 °Z, jeżeli w rurce polarymetrycznej o długości 200 mm znajduje się 26 g cukru w 100 ml wody.
2. Polaryzację oznacza się za pomocą sacharymetru lub polarymetru.
3. Odczynnikami używanymi do oznaczania polaryzacji są:
1) środek klarujący będący roztworem zasadowego octanu ołowiawego;
2) eter etylowy.
4. W celu przygotowania środka klarującego dodaje się 560 g sproszkowanego zasadowego octanu ołowiawego do około 1 000 ml świeżo zagotowanej destylowanej wody, a następnie mieszaninę tę gotuje się przez 30 minut i pozostawia na noc. W celu uzyskania roztworu o gęstości d = 1,25 g/ml, w temperaturze 20 °C, dekantuje się sklarowaną nad osadem ciecz i rozcieńcza świeżo zagotowaną wodą. Roztwór zabezpiecza się przed kontaktem z powietrzem.
5. Sprzęt laboratoryjny służący do oznaczania polaryzacji stanowią:
1) polarymetr z Międzynarodową Skalą Cukrową wyrażoną w stopniach sacharymetrycznych (°Z) i dokładnością odczytu 0,01 °Z lub inny polarymetr z podziałką liniową;
2) lampa sodowa;
3) rurki polarymetryczne o długości 200 mm z odchyleniem ą 0,02 mm;
4) waga analityczna o dokładności 0,1 mg;
5) wzorcowane 100 ml kolby miarowe;
6) łaźnia wodna z termoregulacją, o temperaturze 20 ą 0,1 °C.
6. Aparat instaluje się w pomieszczeniu umożliwiającym utrzymanie stałej temperatury 20 °C. Przyrząd wzorcuje się za pomocą standardowych płytek kwarcowych.
W przypadku stosowania polarymetru z podziałką liniową, w celu uzyskania wyników w Międzynarodowej Skali Cukrowej, uzyskane wartości przelicza się na stopnie sacharymetryczne °Z.
Kolby o rzeczywistej pojemności w zakresie 100 ą 0,01 ml stosuje się bez korekty. Kolby o większej pojemności są odpowiednio korygowane.
7. Przygotowanie roztworu
Odważa się szybko 26 ą 0,002 g badanej próbki i przenosi się ją do kolby miarowej o pojemności 100 ml, zawierającej mniej więcej 60 ml wody. Następnie rozpuszcza się cukier, obracając kolbą, bez podgrzewania. W razie konieczności klarowania dodaje się 0,5 ml odczynnika octanu ołowiawego. Następnie miesza się zawartość kolby ruchem obrotowym i dopełnia wodą do menisku znajdującego się mniej więcej 10 mm poniżej kreski. Kolbę umieszcza się w łaźni wodnej, utrzymując temperaturę 20 ą 0,1 °C aż do uzyskania temperatury roztworu cukru takiej jak w łaźni.
Za pomocą kropli eteru etylowego usuwa się wszystkie pęcherzyki powstałe na powierzchni cieczy. Dopełnia się wodą do kreski. Zamyka się kolbę i miesza dokładnie, odwracając co najmniej trzykrotnie kolbę do góry dnem, a następnie pozostawia się ją na 5 minut.
8. Wykonanie oznaczenia
Próbkę filtruje się przez bibułę, następnie odrzuca się pierwsze 10 ml i zbiera się następne 50 ml przesączu. Płucze się rurkę polarymetryczną dwukrotnie badanym roztworem, po czym napełnia się rurkę polarymetryczną badanym roztworem w temperaturze 20 ą 0,1 °C, a następnie usuwa się wszystkie pęcherzyki powietrza i umieszcza się napełnioną rurkę w sacharymetrze lub polarymetrze. Mierzy się stopnie sacharymetryczne lub stopnie kątowe, dokonując pomiaru trzykrotnie, i oblicza się średnią. Podczas wykonywania wszystkich kolejnych czynności utrzymuje się temperaturę 20 ą 1 °C.
9. Wyrażanie wyników
Wyniki wyraża się w stopniach sacharymetrycznych °Z, z dokładnością do 0,1 °Z, z uwzględnieniem powtarzalności wyników. W celu przekształcenia stopni kątowych na stopnie sacharymetryczne °Z stosuje się następujący wzór:
°Z = stopień kątowy 2,889
Przyjmuje się, że różnica między wynikami dwóch oznaczeń, przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach – przy czym oba oznaczenia stanowią średnią z pięciu odczytów – nie powinna przekraczać 0,1 °Z.
Załącznik nr 2
METODA OZNACZANIA ZAWARTOŚCI CUKRU INWERTOWANEGO W CUKRZE PRZEMYSŁOWYM – METODA INSTYTUTU BERLIŃSKIEGO
1. Oznaczanie zawartości cukru inwertowanego według Instytutu Berlińskiego polega na redukcji kompleksów miedzi II przez cukry inwertowane zawarte w roztworze, na skutek czego powstaje tlenek miedziowy l, który jest następnie utleniany za pomocą wzorcowego roztworu jodu, a jego nadmiar jest oznaczany przez miareczkowanie mianowanym roztworem tiosiarczanu sodowego.
2. Odczynnikami używanymi do oznaczania cukrów redukujących są:
1) roztwór miedzi II (roztwór Müllera);
2) roztwór kwasu octowego o stężeniu 5 mol/litr;
3) roztwór jodu o stężeniu 0,0333 N;
4) roztwór tiosiarczanu sodowego o stężeniu 0,0333 mol/litr;
5) roztwór skrobi.
3. W celu przygotowania roztworu miedzi II rozpuszcza się 35 g pięciowodnego siarczanu miedzi II (CuSO4 x 5 H2O) w 400 ml gotującej się wody i pozostawia się do ostudzenia. Następnie rozpuszcza się 173 g czterowodnego winianu sodowo-potasowego (sól Rochelle lub sól Seignette; KNaC4H4O6 x 4 H2O) oraz 68 g bezwodnego węglanu sodowego (Na2CO3) w 500 ml gotującej się wody i pozostawia się do ostudzenia.
Otrzymane dwa roztwory przenosi się do kolby o pojemności 1 000 ml i uzupełnia wodą do kreski. Następnie dodaje się 2 g węgla aktywnego, miesza się, pozostawia na kilka godzin i filtruje przez gęstą bibułę lub membranę filtracyjną.
Jeżeli podczas przechowywania pojawiają się niewielkie ilości tlenku miedzi l, roztwór filtruje się ponownie.
4. W celu otrzymania roztworu jodu o stężeniu 0,0333 N rozpuszcza się 4,2258 g jodu w 1 000 ml wody.
5. W celu otrzymania roztworu skrobi dodaje się do 1 000 ml wrzącej wody 5 g rozpuszczalnej skrobi, rozmieszanej wcześniej w 30 ml wody, i utrzymuje się wrzenie przez 3 minuty. Następnie pozostawia się do ostudzenia i dodaje, w miarę potrzeb, 10 mg jodku rtęci II jako środka konserwującego.
6. Sprzęt laboratoryjny służący do oznaczania cukrów redukujących stanowią:
1) kolba stożkowa o pojemności 300 ml;
2) dokładne biurety i pipety;
3) łaźnia wodna, wrząca.
7. Przygotowanie roztworu
W celu przygotowania roztworu odważa się 10 g lub mniej badanej próbki do 300 ml kolby stożkowej i rozpuszcza się ją w 100 ml wody. Próbka powinna zawierać nie więcej niż 30 mg cukru inwertowanego.
8. Wykonanie oznaczenia
Odmierza się pipetą 10 ml roztworu miedzi II do kolby z badaną próbką, miesza się zawartość, i umieszcza kolbę we wrzącej łaźni na 10 minut. Poziom roztworu w kolbie stożkowej powinien być co najmniej 20 mm poniżej poziomu wody w łaźni wodnej.
Po 10 minutach szybko studzi się kolbę pod bieżącym strumieniem zimnej wody, nie mieszając roztworu, ponieważ tlen atmosferyczny mógłby ponownie utlenić część wytrąconego tlenku miedzi l.
Kolejno za pomocą pipety dodaje się 5 ml kwasu octowego o stężeniu 5 mol/litr bez mieszania, szybko dodaje z biurety nadmiar (między 20 a 40 ml) roztworu jodu o stężeniu 0,0333 N i miesza się aż do rozpuszczenia osadu miedzi. Następnie odmiareczkowuje się nadmiar jodu roztworem tiosiarczanu sodowego o stężeniu 0,0333 mol/litr, stosując jako wskaźnik roztwór skrobi, dodany pod koniec miareczkowania.
9. Wykonanie próby ślepej
Próbę ślepą wykonuje się dla każdego nowego roztworu miedzi II, zastępując badaną próbkę cukru wodą, przy założeniu, że miareczkowanie nie powinno przekraczać 0,1 ml.
10. Wykonanie próby kontrolnej na zimno
W celu wykonania na zimno próby kontrolnej z roztworem cukru pozostawia się ją w temperaturze pokojowej na 10 minut, aby pozwolić na ewentualną reakcję obecnych czynników redukujących, np. dwutlenku siarki.
11. Wyrażanie wyników
Wyniki wyrażane są w procentach zawartości cukru inwertowanego, z uwzględnieniem powtarzalności wyników.
Przyjmuje się, że każdy mililitr roztworu jodu, który brał udział w reakcji, odpowiada 1 mg cukru inwertowanego, a objętość zużytego jodu odpowiada, po dokonaniu korekty, mililitrom 0,0333 N jodu dodanego w nadmiarze po odjęciu ilości (w mililitrach) tiosiarczanu sodowego o stężeniu 0,0333 mol/litr użytego do miareczkowania.
W celu skorygowania objętości (w mililitrach) zużytego 0,0333 N jodu odejmuje się:
1) liczbę mililitrów zużytych w próbie ślepej przeprowadzonej z wodą;
2) liczbę mililitrów zużytych na zimno w próbie z roztworem cukru;
3) 2 ml z tytułu 10 g cukru obecnego w stosowanej próbie (lub jej wielokrotność) lub proporcjonalną ilość, gdy próbka zawiera mniej niż 10 g cukru (korekta na cukier).
Zawartość cukru inwertowanego (Inw) w procentach oblicza się według wzoru:
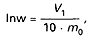
gdzie:
V1 – oznacza ilość roztworu jodu po korekcie, w mililitrach,
mo – oznacza masę zastosowanej próbki, w gramach.
Przyjmuje się, że różnica między wynikami dwóch oznaczeń przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 0,02 g na 100 g próbki.
Załącznik nr 3
METODA OZNACZANIA ZAWARTOŚCI CUKRU INWERTOWANEGO W CUKRZE (CUKRZE BIAŁYM) I W CUKRZE EKSTRABIAŁYM (CUKRZE RAFINOWANYM) – METODA KNIGHTA I ALLENA
1. Oznaczanie zawartości cukru inwertowanego metodą Knighta i Allena polega na dodaniu do roztworu próbki odczynnika miedzi II i odmiareczkowaniu roztworem EDTA jego nadmiaru przed redukcją i po niej w badanej próbce.
2. Odczynnikami używanymi do oznaczania zawartości cukru inwertowanego metodą Knighta i Allena są:
1) roztwór wersenianu dwusodowego (EDTA) o stężeniu 0,0025 mol/litr;
2) roztwór wskaźnika mureksydowego;
3) odczynnik alkaliczny miedzi;
4) wzorcowy roztwór cukru inwertowanego;
5) czysta sacharoza.
3. W celu otrzymania roztworu wersenianu dwusodowego (EDTA) rozpuszcza się 0,93 g EDTA w wodzie i uzupełnia wodą do 1 000 ml.
4. W celu otrzymania roztworu wskaźnika mureksydowego dodaje się 0,25 g mureksydu do 50 ml wody i miesza z 20 ml 0,2 g/100 ml wodnego roztworu błękitu metylenowego.
5. W celu otrzymania odczynnika alkalicznego miedzi rozpuszcza się 25 g bezwodnego węglanu sodowego i 25 g czterowodnego winianu potasowo-sodowego w około 600 ml wody zawierającej 40 ml wodorotlenku sodu o stężeniu 1 mol/litr. Następnie rozpuszcza się 6 g pięciowodnego siarczanu miedzi II (CuSO4 x 5 H2O) w około 100 ml wody, dodaje do roztworu winianu i uzupełnia się wodą do objętości 1 000 ml. Roztwór jest trwały około 7 dni.
6. W celu otrzymania wzorcowego roztworu cukru inwertowanego rozpuszcza się 23,75 g czystej sacharozy w około 120 ml wody w kolbie z podziałką o pojemności 250 ml; dodaje się 9 ml kwasu solnego (d = 1,16 g/ml) i pozostawia w temperaturze pokojowej na 8 dni. Następnie uzupełnia się roztwór wodą do 250 ml i sprawdza stopień przeprowadzonej hydrolizy za pomocą odczytu polarymetrycznego lub sacharymetrycznego w rurce 200 mm. Odczyt powinien wynosić 11,80 ą 0,05 °Z. Odmierza się pipetą 200 ml tego roztworu do kolby o pojemności 2 000 ml z jedną kreską. Rozcieńcza się wodą i wstrząsając (aby uniknąć przeanalizowania miejscowego), dodaje się 71,4 ml roztworu wodorotlenku sodowego (1 ml/litr), w którym rozpuszczono 4 g kwasu benzoesowego. Dopełnia się wodą do 2 000 ml, aby otrzymać roztwór 1 g/100 ml cukru inwertowanego. Roztwór ten powinien wykazywać pH ≈ 3.
Trwały roztwór podstawowy należy rozcieńczać dopiero bezpośrednio przed użyciem.
Czysta sacharoza jest próbką zawierającą czystą sacharozę oraz cukier inwertowany w ilości nie większej niż 0,001 g/100 g.
7. Sprzęt laboratoryjny służący do oznaczania cukru inwertowanego metodą Knighta i Allena stanowią:
1) probówki, o wymiarach 150 x 20 mm;
2) parownica porcelanowa, biała;
3) waga analityczna, o dokładności 0,1 mg.
8. Przygotowanie roztworu
W celu przygotowania roztworu w probówce rozpuszcza się 5 g próbki cukru w 5 ml zimnej wody, dodaje się 2 ml odczynnika alkalicznego miedzi, miesza i zanurza się probówkę na 5 minut we wrzącej łaźni wodnej, a następnie studzi w zimnej wodzie.
9. Wykonanie oznaczenia
Przenosi się ilościowo roztwór z probówki do parownicy porcelanowej, używając możliwie jak najmniejszej ilości wody, dodaje 3 krople wskaźnika mureksydowego i miareczkuje roztworem EDTA, a następnie odczytuje wynik w mililitrach EDTA zużytego do miareczkowania (oznaczony jako Vo.).
Pod koniec zabarwienie roztworu zmienia się z zielonego poprzez szary, aż do końcowego purpurowego. Barwa purpurowa powoli znika z powodu utleniania się tlenku miedzi l do tlenku miedzi II, zależnie od stężenia miedzi zredukowanej, stąd miareczkowanie należy wykonać stosunkowo szybko.
10. Sporządzenie wykresu kalibrowania
W celu sporządzenia wykresu kalibrowania dodaje się znane ilości cukru inwertowanego (wzorcowy roztwór cukru inwertowanego odpowiednio rozcieńczony) do 5 g czystej sacharozy oraz odpowiedniej ilości zimnej wody – tak, aby łącznie dodać 5 ml roztworu, a następnie wykreśla się zależność zużytych mililitrów EDTA podczas miareczkowania (w mililitrach) w stosunku do procentowej ilości cukru inwertowanego, dodanego do 5 g sacharozy. Otrzymany wykres jest linią prostą w zakresie 0,001 do 0,019 g/100 g cukru inwertowanego/100 g próbki.
11. Wyrażanie wyników
Wyniki wyrażane są w procentach zawartości cukru inwertowanego, z uwzględnieniem powtarzalności wyników.
Na wykresie kalibracyjnym odczytuje się procent zawartości cukru inwertowanego, odpowiadający wartości Vo ml EDTA, oznaczonej podczas badania próbki.
Jeżeli w analizowanej próbce stężenie cukru inwertowanego na 100 g próbki jest większe niż 0,017 g, to zmniejsza się odpowiednio ilość próbki pobieranej i dopełnia się badaną próbkę do 5 g czystą sacharozą.
W celu przeliczenia stopni sacharymetrycznych °Z na polarymetryczne stopnie kątowe należy stopnie sacharymetryczne °Z podzielić przez 2,889.
Przyjmuje się, ze różnica między wynikami dwóch oznaczeń przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 0,005 g na 100 g próbki.
Załącznik nr 4
METODA l OZNACZANIA ZAWARTOŚCI CUKRÓW REDUKUJĄCYCH W PŁYNNYM CUKRZE (ROZTWORZE CUKRU), W PŁYNNYM CUKRZE INWERTOWANYM (ROZTWORZE CUKRU INWERTOWANEGO) l W SYROPIE CUKRU INWERTOWANEGO WYRAŻONYCH JAKO RÓWNOWAŻNIK CUKRU INWERTOWANEGO LUB DEKSTROZY (D-GLUKOZY) – METODA LUFF-SCHOORLA
1. Oznaczanie zawartości cukrów redukujących metodą Luff-Schoorla polega na podgrzaniu badanej próbki do temperatury wrzenia w warunkach standardowych z roztworem miedzi II, która zostaje częściowo zredukowana do miedzi l. Nadmiar miedzi II oznacza się metodą jodometryczną.
2. Odczynnikami do oznaczania cukrów redukujących metodą Luff-Schoorla są:
1) roztwór Carreza l;
2) roztwór Carreza II;
3) odczynnik Luff-Schoorla;
4) roztwór tiosiarczanu sodowego o stężeniu 0,1 mol/litr;
5) roztwór skrobi;
6) kwas siarkowy o stężeniu 3 mol/litr;
7) roztwór jodku potasowego 30% (m/v);
8) wiórki pumeksowe, gotowane w kwasie solnym, przepłukane wodą i osuszone;
9) izopentanol;
10) wodorotlenek sodowy o stężeniu 0,1 mol/litr;
11) kwas solny o stężeniu 0,1 mol/litr;
12) roztwór fenoloftaleiny, 1% (m/v) w etanolu.
3. W celu przygotowania roztworu Carreza l dodaje się do 21,95 g dwuwodnego octanu cynkowego (Zn(CH3COO)2 x 2 H2O) lub 24 g trójwodnego octanu cynkowego (Zn(CH3COO)2 x 3 H2O) 3 ml kwasu octowego lodowatego, a następnie dopełnia wodą do 100 ml.
4. W celu przygotowania roztworu Carreza II rozpuszcza się 10,6 g trójwodnego sześciocyjano-żelazianu potasowego II K4[Fe(CN)6] x 3 H2O w wodzie i dopełnia wodą do 100 ml.
5. W celu przygotowania odczynnika Luff-Schoorla sporządza się roztwory:
1) siarczanu miedzi II;
2) kwasu cytrynowego;
3) węglanu sodowego.
6. W celu sporządzenia roztworu siarczanu miedzi II rozpuszcza się 25 g pięciowodnego bezżelazowego siarczanu miedzi II (CuSO4 x 5 H2O) w 100 ml wody.
7. W celu sporządzenia roztworu kwasu cytrynowego rozpuszcza się 50 g jednowodnego kwasu cytrynowego (C6H8O7 x H2O) w 50 ml wody.
8. W celu sporządzenia roztworu węglanu sodowego rozpuszcza się 143,8 g bezwodnego węglanu sodu (Na2CO3) w około 300 ml ciepłej wody i pozostawia do ostudzenia.
Następnie dodaje się do kolby o pojemności 1 000 ml roztwór kwasu cytrynowego i roztwór węglanu sodowego, ostrożnie miesza się ruchem obrotowym, aż do zaniku piany, i dodaje roztwór siarczanu miedzi II, uzupełniając wodą do 1 000 ml.
Roztwór pozostawia się na 24 godziny, w razie potrzeby filtruje, i sprawdza się molarność tak uzyskanego odczynnika za pomocą metody opisanej we wzorcowaniu odczynnika Luff-Schoorla (Cu 0,1 mol/litr; Na2CO3 1 mol/litr).
9. W celu przygotowania roztworu skrobi dodaje się do 1 000 ml wrzącej wody 5 g rozpuszczalnej skrobi, namoczonej uprzednio w 30 ml wody, i utrzymuje się wrzenie przez 3 minuty, pozostawia do ostudzenia, a następnie – jeżeli roztwór tego wymaga – dodaje 10 mg jodku rtęci II jako środka konserwującego.
10. Sprzęt laboratoryjny służący do oznaczania cukrów redukujących metodą Luff-Schoorla stanowią:
1) kolba stożkowa o pojemności 300 ml, wyposażona w chłodnicę zwrotną;
2) stoper.
11. Wzorcowanie odczynnika Luff-Schoorla
W celu wykonania wzorcowania odczynnika Luff-Schoorla dodaje się do 25 ml odczynnika Luff-Schoorla 3 g 30% jodku potasowego i 25 ml kwasu siarkowego o stężeniu 3 mol/litr, miareczkuje się 0,1 mol/litr tiosiarczanem sodowym w obecności roztworu skrobi, jako wskaźnika, dodanego pod koniec miareczkowania. Jeżeli objętość zużytego 0,1 mol/litr roztworu tiosiarczanu sodowego nie wynosi dokładnie 25 ml, odczynnik należy przyrządzić ponownie.
Następnie odmierza się pipetą 10 ml odczynnika Luff-Schoorla do kolby pomiarowej o pojemności 100 ml, rozcieńcza wodą, pipetą pobiera się 10 ml rozcieńczonego odczynnika i dodaje się do 25 ml 0,1 mol/litr kwasu solnego w kolbie stożkowej. Ogrzewa się przez 1 godzinę we wrzącej łaźni wodnej, studzi się, dopełnia do początkowej objętości świeżo zagotowaną wodą, a następnie miareczkuje 0,1 mol/litr wodorotlenkiem sodowym wobec fenoloftaleiny jako wskaźnika. Zużycie 0,1 mol/litr wodorotlenku sodowego powinno wynosić między 5,5 a 6,5 ml.
Kolejno miareczkuje się 10 ml rozcieńczonego odczynnika 0,1 mol/litr kwasem solnym wobec fenoloftaleiny jako wskaźnika. Punkt końcowy charakteryzuje się zanikiem purpurowego zabarwienia. Zużycie 0,1 mol/litr kwasu solnego powinno wynosić między 6 a 7,5 ml.
Odczyn pH roztworu Luff-Schoorla waha się między 9,3 a 9,4 w temperaturze 20 °C.
12. Przygotowanie roztworu
W celu przygotowania roztworu dokładnie odważa się (z dokładnością do 1 mg) 5 g badanej próbki, przenosi ją ilościowo do kolby o pojemności 250 ml zawierającej 200 ml wody, klaruje – jeżeli roztwór tego wymaga – przez dodanie 5 ml roztworu Carreza l, a następnie 5 ml roztworu Carreza II. Po dodaniu obu roztworów dokładnie miesza się, uzupełnia wodą do 250 ml, dobrze miesza. Jeżeli to konieczne, filtruje się.
Następnie rozcieńcza się roztwór tak, aby 25 ml roztworu zawierało nie mniej niż 15 mg i nie więcej niż 60 mg cukrów redukujących, wyrażonych jako glukoza.
13. Miareczkowanie metodą Luff-Schoorla
W celu wykonania miareczkowania metodą Luff-Schoorla do kolby stożkowej o pojemności 300 ml pobiera się pipetą 25 ml odczynnika Luff-Schoorla, dodaje się pipetą 25 ml roztworu cukru i umieszcza w niej 2 wiórki pumeksowe. Kolbę łączy się z chłodnicą zwrotną i całość szybko umieszcza na siatce azbestowej nad palnikiem Bunsena. Siatka powinna mieć otwór wycięty w części azbestowej o tej samej średnicy co podstawa kolby. Utrzymuje się ciecz w temperaturze wrzenia przez mniej więcej 2 minuty, ogrzewa na wolnym ogniu dokładnie przez 10 minut, następnie studzi się próbkę w zimnej wodzie, a po 5 minutach dodaje się 10 ml roztworu jodku potasowego i szybko, z zachowaniem ostrożności (ze względu na burzliwy przebieg), dodaje 25 ml 3 mol/litr kwasu siarkowego. Całość miareczkuje się za pomocą 0,1 mol/litr roztworu tiosiarczanu sodowego aż do uzyskania prawie bezbarwnego roztworu, a następnie dodaje się kilka mililitrów roztworu skrobi jako wskaźnika, po czym kontynuuje miareczkowanie aż do zaniku niebieskiego zabarwienia.
W celu zmniejszenia tworzenia piany, przed zakwaszaniem kwasem siarkowym, można dodać niewielką ilość izopentanolu.
14. Próba kontrolna
Przeprowadza się próbę kontrolną, stosując 25 ml wody zamiast 25 ml roztworu cukru.
15. Wyrażanie wyników
Wyniki są wyrażane w procentach suchej masy (m/m) w przypadku cukru inwertowanego lub D-glukozy, z uwzględnieniem powtarzalności wyników.
Z tabeli odczytuje się – w razie potrzeby interpolując – masę glukozy lub cukru inwertowanego w miligramach, odpowiadającą różnicy między dwoma odczytami miareczkowania, wyrażonymi w mililitrach 0,1 mol/litr siarczanu sodowego.
Przyjmuje się, że różnica między wynikami dwóch oznaczeń przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 0,2 ml.
Zależność zużycia tiosiarczanu sodu o stężeniu 0,1 mol/litr od zawartości cukrów redukujących w metodzie Luff-Schoorla
| Zużycie w mililitrach Na2S2O3 o stężeniu 1 mol/litr | Glukoza, fruktoza, cukry inwertowane C6H12O6 | |
| ml | mg | różnica |
| 1 | 2,4 |
|
| 2 | 4,8 | 2,4 |
| 3 | 7,2 | 2,4 |
| 4 | 9,7 | 2,5 |
| 5 | 12,2 | 2,5 |
| 6 | 14,7 | 2,5 |
| 7 | 17,2 | 2,5 |
| 8 | 19,8 | 2,6 |
| 9 | 22,4 | 2,6 |
| 10 | 25,0 | 2,6 |
| 11 | 27,6 | 2,6 |
| 12 | 30,3 | 2,7 |
| 13 | 33,0 | 2,7 |
| 14 | 35,7 | 2,7 |
| 15 | 38,5 | 2,8 |
| 16 | 41,3 | 2,8 |
| 17 | 44,2 | 2,9 |
| 18 | 47,1 | 2,9 |
| 19 | 50,0 | 2,9 |
| 20 | 53,0 | 3,0 |
| 21 | 56,0 | 3,0 |
| 22 | 59,1 | 3,1 |
| 23 | 62,2 | 3,1 |
METODA II OZNACZANIA CUKRÓW REDUKUJĄCYCH W PŁYNNYM CUKRZE (ROZTWORZE CUKRU), W PŁYNNYM CUKRZE INWERTOWANYM (ROZTWORZE CUKRU INWERTOWANEGO) l W SYROPIE CUKRU INWERTOWANEGO WYRAŻONYCH JAKO CUKIER INWERTOWANY – WEDŁUG MODYFIKACJI OBJĘTOŚCI STAŁEJ LANE'A l EYNONA
1. Oznaczanie cukrów redukujących według modyfikacji objętości stałej Lane'a i Eynona polega na miareczkowaniu roztworu próbki w temperaturze wrzenia w stosunku do określonej objętości roztworu Fehlinga wobec błękitu metylenowego jako wskaźnika.
2. Odczynnikami używanymi do oznaczania zawartości cukrów redukujących według modyfikacji objętości stałej Lane'a i Eynona są:
1) roztwór Fehlinga;
2) roztwór wodorotlenku sodowego o stężeniu 1 mol/litr;
3) wzorcowy roztwór cukru inwertowanego;
4) roztwór błękitu metylenowego.
3. W celu przygotowania roztworu Fehlinga sporządza się:
1) roztwór A;
2) roztwór B.
4. W celu sporządzenia roztworu A rozpuszcza się 69,3 g pięciowodnego siarczanu miedzi II (CuSO4 x 5 H2O) w wodzie i dopełnia się wodą do 1 000 ml.
5. W celu sporządzenia roztworu B rozpuszcza się w wodzie 346 g pięciowodnego winianu sodowo-potasowego (KNaC4 x 5 H2O) wraz ze 100 g wodorotlenku sodowego (NaOH) i dopełnia się wodą do 1 000 ml. Klarowny roztwór dekantuje się znad osadu.
6. Roztwory A i B należy przechowywać w butelkach z ciemnego (brązowego, bursztynowego) szkła.
7. W celu przygotowania wzorcowego roztworu cukru inwertowanego rozpuszcza się 23,75 g czystej sacharozy w około 120 ml wody w kolbie o pojemności 250 ml z podziałką, dodaje się 9 ml kwasu solnego (d = 1,16 g/cm3) i pozostawia na 8 godzin w temperaturze pokojowej. Następnie dopełnia się roztwór do 250 ml i sprawdza się zakończenie hydrolizy za pomocą polarymetru lub sacharymetru przez dokonanie odczytu w rurce polarymetrycznej o długości 200 mm. Odczyt powinien wynieść (11,80 ą 0,05) °Z. Do kolby o pojemności 2 000 ml z podziałką odmierza się pipetą 200 ml tego roztworu. Rozcieńcza się wodą i wstrząsając (aby uniknąć przeanalizowania miejscowego), dodaje się 71,4 ml roztworu wodorotlenku sodowego o stężeniu 1 mol/litr, w którym rozpuszczono 4 g kwasu benzoesowego, dopełnia się wodą do 2 000 ml, aby otrzymać roztwór cukru inwertowanego 1 g/100 ml. Odczyn tego roztworu powinien wynosić pH ≈ 3.
Roztwór podstawowy (trwały) należy rozcieńczać bezpośrednio przed użyciem.
8. W celu uzyskania rozcieńczonego roztworu cukru inwertowanego 0,25 g/100 ml należy 1 g podstawowego roztworu rozcieńczyć wodą do 250 ml w kolbie pomiarowej.
Całość przenosi się do kolby o pojemności 1 000 ml z podziałką i ponownie rozcieńcza wodą do kreski w temperaturze 20 °C.
9. W celu sporządzenia roztworu błękitu metylenowego 1 g błękitu rozcieńcza się wodą w kolbie, a następnie dopełnia wodą do 100 ml.
10. Sprzęt laboratoryjny służący do oznaczania zawartości cukrów redukujących według modyfikacji objętości stałej Lane'a i Eynona stanowią:
1) laboratoryjne kolby żaroodporne z wąską szyjką, o pojemności 500 ml;
2) biureta o pojemności 50 ml z podziałką co 0,05 ml;
3) pipety z podziałką co 20, 25 i 50 ml;
4) kolby pomiarowe z 1 kreską o pojemności 250, 1 000 i 2 000 ml;
5) urządzenie do podgrzewania umożliwiające utrzymanie temperatury wrzenia, zgodnie z warunkami opisanymi we wzorcowaniu roztworu Fehlinga, i pozwalające na obserwacje końcowej zmiany zabarwienia, bez konieczności wyjmowania kolby znad ogrzewacza;
6) stoper wskazujący z dokładnością co najmniej do sekundy.
11. Wzorcowanie roztworu Fehlinga
W celu wykonania wzorcowania do czystej, suchej zlewki pobiera się pipetą 50 ml roztworu B i 50 ml roztworu A, po czym dokładnie miesza jej zawartość. Następnie płucze się i napełnia biuretę 0,25% (0,25 g/100 ml) wzorcowym roztworem cukru inwertowanego.
Do kolby o pojemności 500 ml odmierza się pipetą wielokrotność 20 ml zmieszanych roztworów A i B, dodaje się 15 ml wody, wprowadza się biuretą 39 ml roztworu cukru inwertowanego, wiórek pumeksu i miesza, ostrożnie obracając kolbą.
Podgrzewa się kolbę z zawartością do temperatury wrzenia, utrzymuje tę temperaturę dokładnie przez 2 minuty (w trakcie kolejnych wykonywanych czynności nie wolno zdejmować kolby z ogrzewacza ani dopuścić, aby roztwór przestał wrzeć) i pod koniec dwuminutowego wrzenia dodaje się 3 lub 4 krople roztworu błękitu metylenowego. Roztwór powinien wykazywać intensywne niebieskie zabarwienie.
Następnie dodaje się, za pomocą biurety, wzorcowy roztwór cukru inwertowanego w małych porcjach – początkowo po 0,2 ml, a na końcu pojedyncze krople – aż do osiągnięcia punktu końcowego. Punkt ten wskazuje zanik niebieskiego zabarwienia. Roztwór przyjmuje barwę czerwonawą związaną z powstaniem zawiesiny tlenku miedzi l.
Punkt końcowy powinien być osiągnięty pod koniec 3 minut od momentu zagotowania się roztworu. Końcowe miano V0 powinno wynosić od 39 do 41 ml. Jeżeli V0 jest większe, należy wyrównać stężenie miedzi w roztworze A i powtórzyć proces kalibrowania.
12. Przygotowanie roztworów próbek
Stężenie próbki badanej powinno być takie, aby jej roztwór zawierał od 250 do 400 mg cukru inwertowanego w 100 ml.
13. Badanie wstępne
Badanie wstępne przeprowadza się w celu upewnienia się, że ilość wody dodawanej do 20 ml zmieszanych roztworów A i B jest wystarczająca do uzyskania ostatecznej objętości 75 ml po miareczkowaniu. W tym celu przygotowuje się roztwór próbki do badania: 25 ml roztworu próbki wprowadza się biuretą do kolby, dodaje się 15 ml wody, pozostawia roztwór w stanie wrzenia przez 2 minuty i miareczkuje aż do osiągnięcia punktu końcowego. Następnie do kolby o pojemności 500 ml odmierza się pipetą wielokrotność 20 ml zmieszanych roztworów A i B, dodaje 15 ml wody, wprowadza biureta 39 ml roztworu badanej próbki, wiórek pumeksu i miesza, ostrożnie obracając kolbą.
Podgrzewa się kolbę z zawartością do temperatury wrzenia, utrzymuje tę temperaturę dokładnie przez 2 minuty (w trakcie kolejnych wykonywanych czynności nie wolno zdejmować kolby z ogrzewacza ani dopuścić, aby roztwór przestał wrzeć) i pod koniec dwuminutowego wrzenia dodaje się 3 lub 4 krople roztworu błękitu metylenowego. Roztwór powinien wykazywać intensywne niebieskie zabarwienie.
Jeżeli po dodaniu roztworu błękitu metylenowego utrzymuje się nadal czerwone zabarwienie, to stężenie w użytej próbce jest zbyt duże. W takim przypadku próba jest nieważna i należy powtórzyć badania, stosując mniej stężone roztwory badanej próbki.
Jeżeli w celu uzyskania czerwonego zabarwienia potrzeba więcej niż 50 ml roztworu, należy stosować bardziej stężone roztwory próbki.
Następnie oblicza się ilość wody, jaką należy dodać – przez odjęcie objętości zmieszanego roztworu Fehlinga (20 ml) i roztworu próbki od objętości 75 ml.
14. Końcowa analiza roztworu próbki
W celu przeprowadzenia końcowej analizy roztworu badanej próbki do kolby żaroodpornej pobiera się pipetą 20 ml zmieszanych roztworów Fehlinga i ilość wody wyznaczoną w badaniu wstępnym, następnie dodaje się za pomocą biurety przybliżoną ilość, pomniejszoną o 1 ml, roztworu próbki z badania wstępnego i kawałki pumeksu, miesza się ostrożnie zawartość, obracając kolbą, doprowadza się do wrzenia i miareczkuje aż do osiągnięcia punktu końcowego. Punkt końcowy należy osiągnąć po upływie 1 minuty od momentu dodania roztworu błękitu metylenowego. Końcowe miano = V1.
15. Wyrażanie wyników
Wyniki są wyrażane w miligramach cukru inwertowanego, z uwzględnieniem powtarzalności wyników.
Zawartość cukrów redukujących w próbce wyrażonych jako cukier inwertowany (Inw) oblicza się według wzoru:
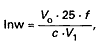
gdzie:
| c | – oznacza stężenie roztworu badanej próbki, w gramach na 100 ml, |
| Vo | – oznacza objętość wzorcowego roztworu cukru inwertowanego, zastosowanego do kalibracyjnego miareczkowania, w mililitrach, |
| V1 | – oznacza objętość roztworu badanej próbki, w mililitrach, |
| f | – oznacza współczynnik korekcji uwzględniający stężenie sacharozy w badanym roztworze próbki. |
Zależność cukru i współczynnika f zestawiono w poniższej tabeli:
| Ilość cukru (g) we wrzącej mieszaninie | Współczynnik korekcji f |
| 0 | 1,000 |
| 0,5 | 0,982 |
| 1,0 | 0,971 |
| 1,5 | 0,962 |
| 2,0 | 0,954 |
| 2,5 | 0,946 |
| 3,0 | 0,939 |
| 3,5 | 0,932 |
| 4,0 | 0,926 |
| 4,5 | 0,920 |
| 5,0 | 0,915 |
| 5,5 | 0,910 |
| 6,0 | 0,904 |
| 6,5 | 0,898 |
| 7,0 | 0,893 |
| 7,5 | 0,888 |
| 8,0 | 0,883 |
| 8,5 | 0,878 |
| 9,0 | 0,874 |
| 9,5 | 0,869 |
| 10,0 | 0,640 |
Korektę innych zawartości sacharozy w badanym roztworze próbki oblicza się z tabeli metodą interpolacji.
Przybliżone stężenie sacharozy można obliczyć, odejmując stężenie substancji rozpuszczalnych związanych z cukrem inwertowanym (do celów obliczenia przyjmuje się f = 1), od ogólnego stężenia rozpuszczonych ciał stałych, wyrażonego jako sacharoza, uzyskane na podstawie wskaźnika załamania roztworu za pomocą metody refraktometrycznej, określonej w załączniku nr 7 do rozporządzenia.
W celu przeliczenia stopni sacharymetrycznych °Z na polarymetryczne stopnie kątowe należy °Z podzielić przez 2,889.
Przyjmuje się, że różnica między wynikami dwóch oznaczeń przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 1 % ich średniej arytmetycznej.
Załącznik nr 5
METODA OZNACZANIA RÓWNOWAŻNIKA DEKSTROZY W SYROPIE GLUKOZOWYM I W SYROPIE GLUKOZOWYM W PROSZKU ORAZ ZAWARTOŚCI DEKSTROZY (D-GLUKOZY) W JEDNOWODNEJ GLUKOZIE (JEDNOWODNEJ DEKSTROZIE) I W BEZWODNEJ GLUKOZIE (BEZWODNEJ DEKSTROZIE) – METODA LANE'A I EYNONA
1. Oznaczanie równoważnika dekstrozy oraz zawartości dekstrozy metodą Lane'a i Eynona polega na miareczkowaniu badanego roztworu w temperaturze wrzenia – w stosunku do określonej objętości roztworu Fehlinga – w ściśle określonych warunkach, wobec błękitu metylenowego jako wskaźnika.
Równoważnik dekstrozy jest to zdolność redukcji obliczona w procentach masy suchej substancji próbki. Zdolność redukcyjna jest to zawartość cukrów redukujących oznaczona za pomocą niniejszej metody, wyrażona w kategoriach bezwodnej dekstrozy (D-glukozy) oraz obliczona w procentach masy próbki.
2. Odczynnikami używanymi do oznaczania równoważnika dekstrozy oraz zawartości dekstrozy metodą Lane'a i Eynona są:
1) roztwór Fehlinga;
2) bezwodna dekstroza (D-glukoza) (C6H12O6);
3) wzorcowy roztwór dekstrozy o stężen
iu 0,6g/100ml;
4) roztwór błękitu metylenowego o stężeniu 0,1 g/100 ml.
3. W celu przygotowania roztworu Fehlinga przygotowuje się roztwór A i B.
4. W celu sporządzenia roztworu A rozpuszcza się 69,3 g pięciowodnego siarczanu miedzi II (CuSO4 x 5 H2O) w wodzie i dopełnia się wodą do 1000 ml.
5. W celu sporządzenia roztworu B rozpuszcza się w wodzie 346 g pięciowodnego winianu sodowo-potasowego (KNaC4 x 5 H2O) wraz z 100 g wodorotlenku sodowego (NaOH) i dopełnia się wodą do 1 000 ml. Klarowny roztwór dekantuje się znad osadu.
6. Następnie do czystej, suchej zlewki pobiera się pipetą 50 ml roztworu B, a następnie 50 ml roztworu A i dobrze miesza.
Zmieszanych roztworów Fehlinga nie przechowuje się. Codziennie przygotowuje się świeże roztwory i kalibruje się.
Roztwory A i B przechowuje się w butelkach z ciemnego (brązowego, bursztynowego) szkła.
W celu przygotowania bezwodnej dekstrozy przed użyciem suszy się ją przez 4 godziny w suszarce próżniowej w temperaturze 100 ą 1 °C lub niższej oraz wewnętrznym ciśnieniu wynoszącym mniej więcej 10 hPa (103 mbar).
7. W celu przygotowania wzorcowego roztworu dekstrozy odważa się (z dokładnością do 0,1 mg) 0,6 g bezwodnej dekstrozy, rozpuszcza się ją w wodzie, przenosi roztwór ilościowo do kolby o pojemności 100 ml, dopełnia wodą do kreski i miesza.
Codziennie stosuje się świeżo przygotowane roztwory.
8. W celu przygotowania roztworu błękitu metylenowego rozpuszcza się 0,1 g błękitu metylenowego w 100 ml wody.
9. Sprzęt laboratoryjny służący do oznaczania równoważnika dekstrozy oraz zawartości dekstrozy metodą Lane'a i Eynona stanowią:
1) laboratoryjne kolby żaroodporne z wąską szyjką, o pojemności 250 ml;
2) biureta, 50 ml, z podziałką co 0,05 ml;
3) pipety, z podziałką co 25 i 50 ml;
4) kolby o pojemności 100 i 500 ml z jedną kreską;
5) urządzenie do podgrzewania, umożliwiające utrzymanie temperatury wrzenia zgodnie z warunkami opisanymi we wzorcowaniu roztworu Fehlinga i pozwalające na obserwacje końcowej zmiany zabarwienia, bez konieczności wyjmowania kolby znad ogrzewacza;
6) stoper z dokładnością pomiaru co najmniej do 1 sekundy.
10. Wzorcowanie roztworu Fehlinga (miareczkowanie wzorcowe)
Do czystej, suchej kolby żaroodpornej pobiera się pipetą 25 ml roztworu Fehlinga, napełnia się biuretę wzorcowym roztworem dekstrozy, korygując menisk do kreski. Następnie do kolby żaroodpornej wprowadza się biuretą 18 ml wzorcowego roztworu dekstrozy i miesza się zawartość, obracając kolbą.
Umieszcza się kolbę żaroodporną na urządzeniu do podgrzewania, uprzednio wyregulowanym tak, aby wrzenie rozpoczęło się w ciągu 120 ą 15 sekund (podgrzewacza nie reguluje się w trakcie trwania całego procesu miareczkowania), uruchamia się stoper w momencie rozpoczęcia wrzenia i po 120 sekundach wrzenia rozpoczyna się dodawanie do kolby żaroodpornej wzorcowego roztworu dekstrozy – za pomocą biurety, w porcjach 0,5 ml – aż do momentu zaniku zabarwienia błękitu metylenowego, a następnie odnotowuje się ogólną objętość dodanego roztworu wzorcowego dekstrozy, łącznie z przedostatnią dawką 0,5 ml (X ml).
Do czystej, suchej kolby żaroodpornej pobiera się pipetą 25 ml roztworu Fehlinga, napełnia się biuretę wzorcowym roztworem dekstrozy, korygując menisk do kreski. Następnie wprowadza się za pomocą biurety do kolby żaroodpornej objętość wzorcowego roztworu dekstrozy równą X – 0,3 ml, umieszcza się kolbę żaroodporną na urządzeniu do podgrzewania, uprzednio wyregulowanym tak, aby wrzenie rozpoczęło się w ciągu 120 ą 15 sekund (podgrzewacza nie reguluje się w trakcie trwania całego procesu miareczkowania), uruchamia się stoper w momencie rozpoczęcia wrzenia i po 120 sekundach wrzenia rozpoczyna się dodawanie do kolby żaroodpornej wzorcowego roztworu dekstrozy – za pomocą biurety, w porcjach początkowo 0,2 ml, a następnie po kropli – do momentu zaniku zabarwienia błękitu metylenowego. Dodawanie kończy się w ciągu 60 sekund, a łączny czas gotowania nie powinien przekroczyć 180 sekund.
Pod koniec tej czynności czas między dodawaniem kolejnych porcji wzorcowego roztworu dekstrozy powinien wynosić od 10 do 15 sekund.
Konieczne jest trzecie miareczkowanie przez dodanie nieco większej od 0,2 ml odpowiednio dostosowanej, początkowej porcji roztworu wzorcowego dekstrozy.
Odnotowuje się objętość (Vo ml) wzorcowego roztworu dekstrozy użytego do punktu końcowego ostatniego miareczkowania, która powinna mieścić się między 19 a 21 ml wzorcowego roztworu dekstrozy, a jeżeli Vo wykracza poza ten zakres, to odpowiednio dostosowuje się stężenie roztworu Fehlinga i powtarza się proces kalibrowania.
Do codziennego kalibrowania roztworu Fehlinga (ponieważ objętość Vo jest dokładnie znana) jest wymagane jedno miareczkowanie przez dodanie początkowej porcji pomniejszonej o 0,5 ml wzorcowego roztworu dekstrozy.
Od momentu rozpoczęcia wrzenia para wydziela się gwałtownie i w sposób ciągły przez cały czas trwania procesu miareczkowania, zapobiegając w ten sposób w maksymalnym stopniu przedostawaniu się powietrza do środka kolby i w konsekwencji reoksydacji jej zawartości.
Zanik zabarwienia błękitu metylenowego jest najlepiej widoczny podczas obserwacji górnych warstw (i menisku) zawartości kolby miareczkowej, ponieważ są one pozbawione strąconego czerwonego tlenku miedzi I. Zanik zabarwienia łatwiej jest obserwować, stosując bezpośrednie oświetlenie. Pomocny jest biały ekran ustawiony za kolbą.
Podczas oznaczania biuretę należy trzymać z dala od źródła ogrzewania.
Ze względu na czynnik indywidualny każdy analityk powinien przeprowadzić własne wzorcowe miareczkowanie i stosować w obliczeniach własną wartość Vo.
11. Wstępne badanie przygotowanej próbki
Jeżeli zdolność redukująca przygotowanej próbki nie jest znana w przybliżeniu, przeprowadza się wstępne badanie w celu uzyskania przybliżonej wartości po to, aby obliczyć masę próbki pobranej do badania.
W celu wstępnego zbadania próbki przygotowuje się 2 % (m/v) roztwór próbki, mający przybliżoną wartość „Z”, napełnia się nim biuretę i koryguje menisk do kreski. Do czystej, suchej kolby żaroodpornej pobiera się pipetą 25 ml roztworu Fehlinga, wprowadza się biuretą 10 ml 2 % roztworu próbki i umieszcza się kolbę żaroodporną na urządzeniu do podgrzewania, uprzednio wyregulowanym tak, aby wrzenie rozpoczęło się w ciągu 120 ą 15 sekund. Następnie podgrzewa się zawartość kolby do wrzenia, dodając 1 ml roztworu błękitu metylenowego, natychmiast po rozpoczęciu wrzenia uruchamia się stoper i za pomocą biurety rozpoczyna się dodawanie roztworu próbki do kolby porcjami 1 ml, w odstępach mniej więcej 10-sekundowych, aż do zaniku niebieskiego zabarwienia błękitu metylenowego. Odnotowuje się całkowitą objętość dodanego roztworu próbki łącznie z przedostatnią porcją (Y ml).
Wartość „Y” nie może przekraczać 50 ml, a w przypadku jej przekroczenia zwiększa się stężenie roztworu próbki i powtarza się miareczkowanie.
Przybliżoną zdolność redukcyjną (Zred) badanej próbki wyrażoną w procentach (m/m) oblicza się według wzoru:
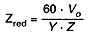
12. Przygotowanie roztworu do badań
Odważa się (z dokładnością do 0,1 mg) próbkę, która zawiera od 2,85 do 3,15 g cukrów redukujących, wyrażonych jako bezwodna dekstroza (D-glukoza), umieszcza się w kolbie miarowej i rozpuszcza się w wodzie, uzupełniając wodą do 500 ml.
Do obliczeń stosuje się znaną w przybliżeniu wartość zdolności redukującej lub przybliżoną wartość zdolności redukcyjnej uzyskaną we wstępnym badaniu przygotowanej próbki.
13. Wykonanie oznaczenia (miareczkowanie oznaczające)
Do czystej, suchej kolby żaroodpornej pobiera się pipetą 25 ml roztworu Fehlinga. Napełnia się biuretę badanym roztworem i wprowadza się go w ilości 18,5 ml do kolby żaroodpornej, mieszając zawartość kolbą. Umieszcza się kolbę żaroodporną na urządzeniu do podgrzewania, uprzednio wyregulowanym tak, aby wrzenie rozpoczęło się w ciągu 120 ą 15 sekund. Następnie podgrzewa się zawartość kolby do wrzenia, dodając 1 ml roztworu błękitu metylenowego. Włącza się stoper i po 120 sekundach wrzenia rozpoczyna się dodawanie biuretą do kolby żaroodpornej roztworu badanej próbki, porcjami po 0,5 ml, aż do momentu zaniku zabarwienia błękitu metylenowego. Odnotowuje się całkowitą objętość dodanego roztworu badanej próbki łącznie z przedostatnią porcją (X ml).
Do czystej, suchej kolby żaroodpornej pobiera się pipetą 25 ml roztworu Fehlinga. Napełnia się biuretę badanym roztworem próbki i wprowadza się go w ilości X – 0,3 ml do kolby żaroodpornej, mieszając zawartość kolbą. Umieszcza się kolbę żaroodporną na urządzeniu do podgrzewania, uprzednio wyregulowanym tak, aby wrzenie rozpoczęło się w ciągu 120 ą 15 sekund. Następnie podgrzewa się zawartość kolby do wrzenia, dodając 1 ml roztworu błękitu metylenowego. Włącza się stoper i po 120 sekundach wrzenia rozpoczyna się dodawanie biuretą do kolby żaroodpornej roztworu badanej próbki, porcjami początkowo po 0,2 ml, a następnie po kropli, aż do momentu zaniku zabarwienia błękitu metylenowego. Dodawanie kończy się w ciągu 60 sekund, a łączny czas gotowania nie powinien przekroczyć 180 sekund.
Pod koniec tej czynności czas między dodawaniem kolejnych porcji roztworu badanej próbki powinien wynosić od 10 do 15 sekund.
Konieczne jest trzecie miareczkowanie przez dodanie nieco większej od 0,2 ml, odpowiednio dostosowanej, początkowej porcji roztworu badanej próbki.
Odnotowuje się objętość (V1) roztworu badanego użytego do punktu końcowego ostatniego miareczkowania, która powinna wynosić od 19 do 21 ml roztworu badanego.
Jeżeli objętość V1 wykracza poza ten zakres, należy odpowiednio dostosować stężenie roztworu badanego i powtórzyć czynności opisane powyżej.
Przeprowadza się dwa oznaczenia z tym samym badanym roztworem.
Zawartość suchej masy w przygotowanej próbce oznacza się metodą określoną w załączniku nr 8 do rozporządzenia.
14. Wyrażanie wyników
Wyniki są wyrażane w procentach masy badanej próbki i masy substancji suchej, z uwzględnieniem powtarzalności wyników.
Zdolność redukcyjną (RP) oblicza się jako procent masy badanej próbki według wzoru:
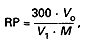
gdzie:
| Vo | – oznacza objętość w mililitrach wzorcowego roztworu dekstrozy użytego do wzorcowania roztworu Fehlinga, |
| V1 | – oznacza objętość w mililitrach roztworu badanej próbki użytego do wykonania oznaczenia, |
| M | – oznacza masę w gramach badanej próbki użytej do sporządzenia 500 ml roztworu badanego. |
Równoważnik dekstrozy (Rd) oblicza się jako procent masy substancji suchej w przygotowanej próbce według wzoru:
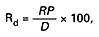
gdzie:
| RP | – oznacza zdolność redukcyjną obliczoną jako procent masy badanej próbki, |
| D | – oznacza zawartość suchej substancji w badanej próbce, w procentach (m/m). |
Jako wynik podaje się średnią arytmetyczną dwóch oznaczeń.
Przyjmuje się, że różnica między wynikami dwóch oznaczeń przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 1 % ich średniej arytmetycznej.
Załącznik nr 6
METODA OZNACZANIA WILGOTNOŚCI CUKRU (CUKRU BIAŁEGO), CUKRU EKSTRABIAŁEGO (CUKRU RAFINOWANEGO), CUKRU PRZEMYSŁOWEGO I FRUKTOZY
1. Oznaczanie wilgotności cukru jako ubytku masy wskutek suszenia polega na wysuszeniu próbki cukru w temperaturze 103 ą 2 °C.
2. Sprzęt laboratoryjny służący do oznaczania wilgotności jako ubytku masy wskutek suszenia stanowią:
1) waga analityczna, o dokładności 0,1 mg;
2) suszarka odpowiednio wentylowana, o płaskim dnie, odpornym na niszczące działanie próbek i na warunki badawcze;
3) naczynka wagowe (aluminiowe, niklowe, szklane), z płaskim dnem, odporne na niszczące działanie próbek i na warunki badawcze, o średnicy od 60 do 100 mm i wysokości od 20 do 30 mm;
4) eksykator, zawierający świeży żel krzemionkowy lub równorzędny środek suszący, ze wskaźnikiem zawartości wody.
3. Suszenie badanej próbki
Wszystkie czynności wykonuje się natychmiast po otwarciu pojemnika z próbką. W celu wykonania suszenia doprowadza się naczynko wagowe do stałej wagi przez suszenie w temperaturze 103 ą 2 °C, pozostawia się je do ochłodzenia w eksykatorze co najmniej na 30–35 minut i waży się z dokładnością do 0,1 mg.
Następnie odważa się do naczynka wagowego (z dokładnością do 0,1 mg) około 20–30 g próbki, umieszcza się naczynko wagowe z próbką w suszarce na okres 3 godzin w temperaturze 103 ą 2 °C, wyjmuje się je i pozostawia do ostudzenia w eksykatorze.
4. Ważenie próbki po ostudzeniu
Waży się próbkę z dokładnością 0,1 mg, ponownie umieszcza się naczynko w suszarce na okres 30 minut w temperaturze 103 ą 2 °C, wyjmuje się, pozostawia się je do ostudzenia w eksykatorze i waży się z dokładnością do 0,1 mg.
Czynność powtarza się, jeżeli różnica między dwoma pomiarami masy wynosi więcej niż 1 mg. W razie pojawienia się przyrostu wagi do obliczeń należy użyć najniższego odnotowanego odczytu.
Przyjmuje się, że łączny czas suszenia nie może przekraczać 4 godzin.
5. Wyrażanie wyników
Wyniki są wyrażane w procentach wilgotności jako strata masy, z uwzględnieniem powtarzalności wyników.
Wilgotność (W) wyrażoną w procentach oblicza się według wzoru:
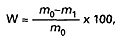
gdzie:
m0 – oznacza masę badanej próbki przed suszeniem, w gramach,
m1 – oznacza masę badanej próbki po suszeniu, w gramach.
Przyjmuje się, że różnica między wynikami dwóch oznaczeń przeprowadzanych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 0,02 g na 100 g próbki.
Załącznik nr 7
METODA OZNACZANIA ZAWARTOŚCI SUCHEJ MASY W PŁYNNYM CUKRZE (ROZTWORZE CUKRU), PŁYNNYM CUKRZE INWERTOWANYM (ROZTWORZE CUKRU INWERTOWANEGO) I W SYROPIE CUKRU INWERTOWANEGO – METODA REFRAKTOMETRYCZNA
1. Oznaczanie zawartości suchej masy w cukrze metodą refraktometryczną polega na oznaczeniu współczynnika załamania roztworu badanej próbki w temperaturze 20 °C i przeliczeniu na zawartość suchej masy za pomocą tabel wskazujących stężenie jako funkcję współczynnika załamania.
2. Aparaturę służącą do oznaczania zawartości suchej masy metodą refraktometryczną stanowią:
1) refraktometr, z dokładnością do czterech miejsc po przecinku, zaopatrzony w termometr i wodną pompę cyrkulacyjną podłączoną do łaźni wodnej z termoregulacją o temperaturze 20 ą 0,5 °C;
2) lampa sodowa.
3. Wykonanie oznaczenia
Polega ono na zmierzeniu za pomocą refraktometru współczynnika załamania próbki w temperaturze 20 °C.
Jeżeli w badanej próbce, przed zmierzeniem, znajdują się kryształki, rozpuszcza się je przez rozcieńczenie 1:1 (m/m).
4. Wyrażanie wyników
Wyniki są wyrażane według zawartości suchej masy, z uwzględnieniem powtarzalności wyników.
Zawartość suchej masy oblicza się według poniższej tabeli na podstawie współczynnika załamania roztworów cukru w temperaturze 20 °C. Wynik koryguje się na obecność cukrów inwertowanych, dodając do otrzymanego z tabeli wyniku 0,022 do każdego procentu inwertowanego cukru obecnego w analizowanej próbce.
Jeżeli próbka była rozcieńczana wodą w stosunku 1:1 (m/m), obliczoną suchą masę mnoży się przez 2.
Przyjmuje się, że różnica między wynikami dwóch oznaczeń przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 0,2 g na 100 g próbki.
TABLICE PRZELICZENIOWE
Współczynniki załamania (n) roztworów cukru w temperaturze 20 °C
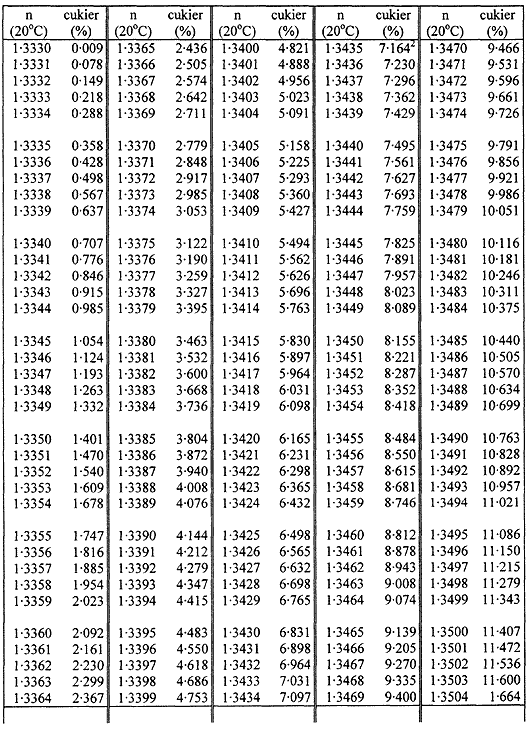
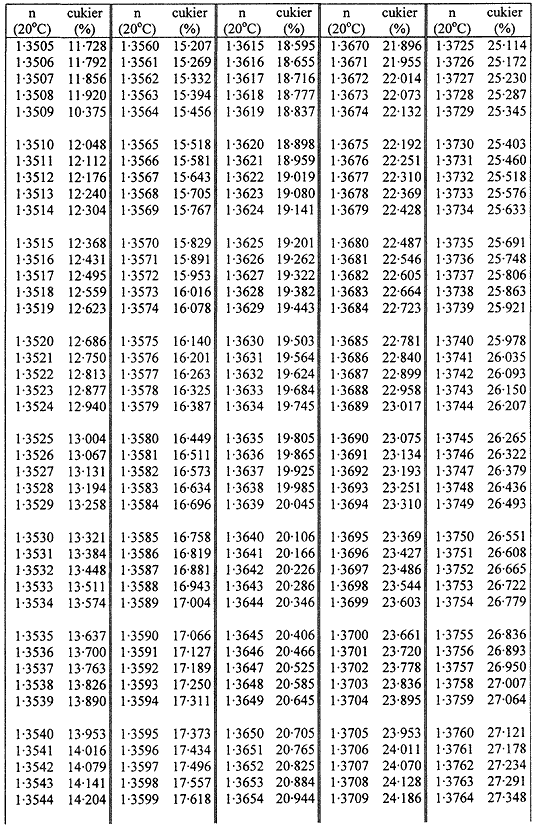
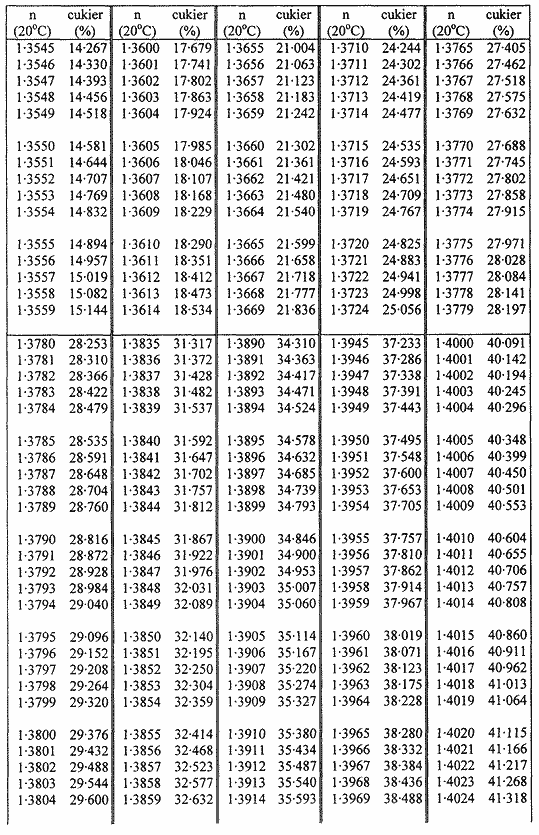
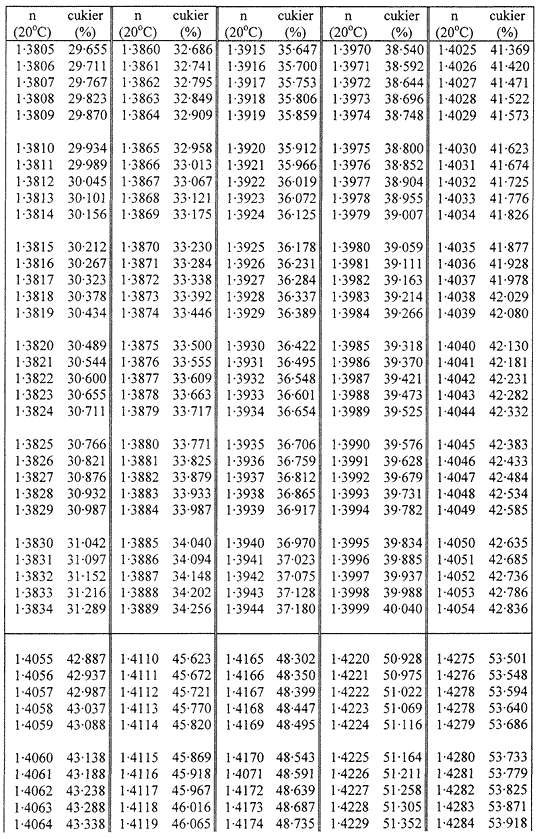
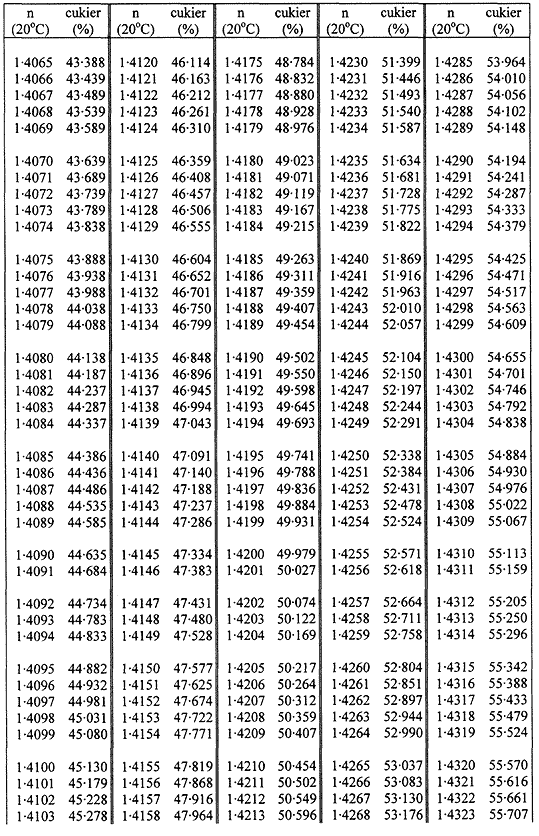
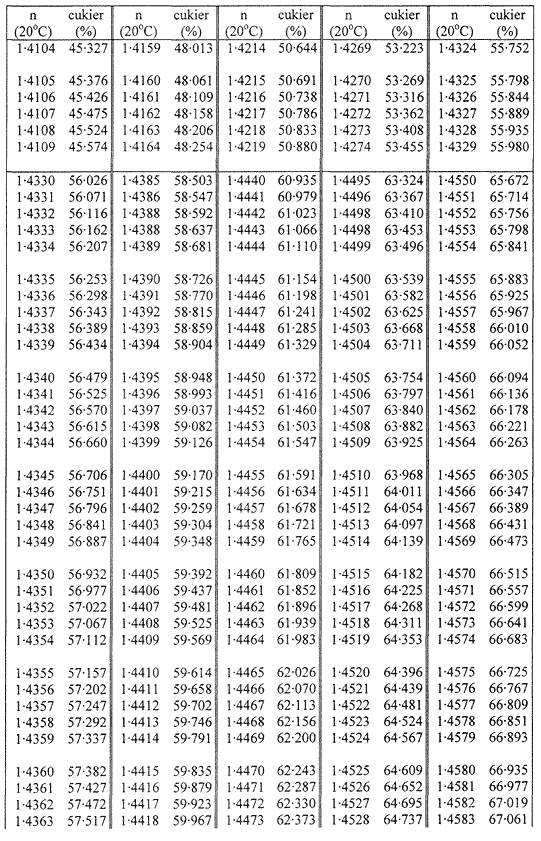
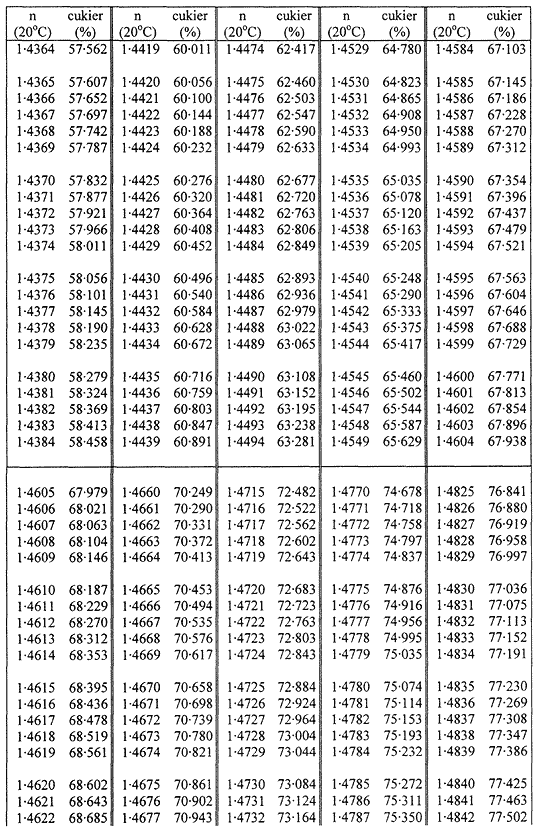
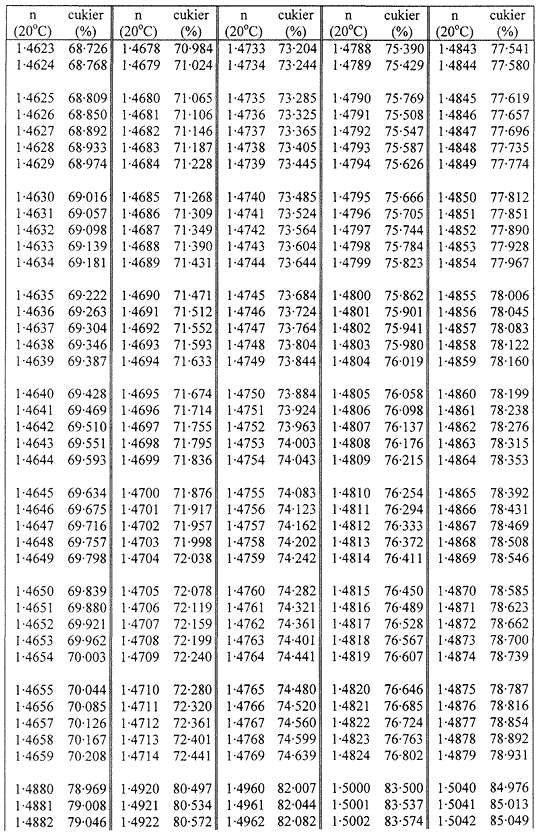
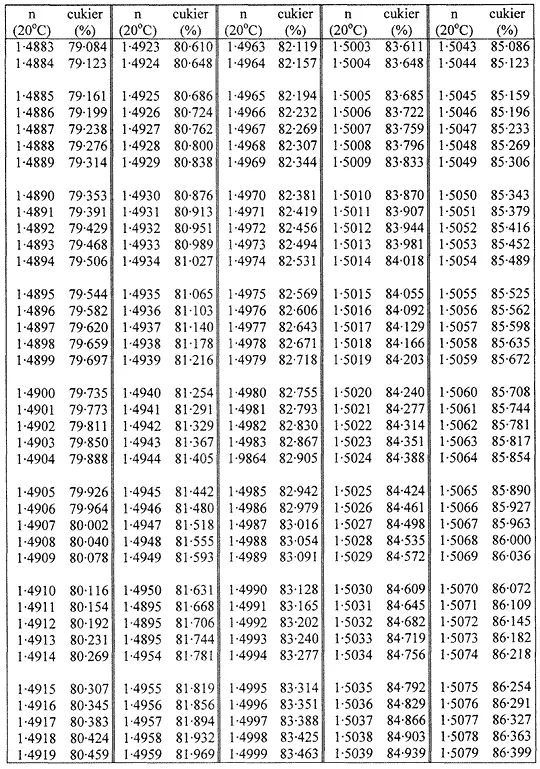
Załącznik nr 8
METODA OZNACZANIA ZAWARTOŚCI SUCHEJ MASY W SYROPIE GLUKOZOWYM, SYROPIE GLUKOZOWYM W PROSZKU, W JEDNOWODNEJ GLUKOZIE (JEDNOWODNEJ DEKSTROZIE) I W BEZWODNEJ GLUKOZIE (BEZWODNEJ DEKSTROZIE) – METODA SUSZARKI PRÓŻNIOWEJ
1. Metoda suszarki próżniowej polega na oznaczeniu suchej masy w temperaturze 70 ą 1 °C za pomocą suszarki próżniowej pod ciśnieniem nieprzekraczającym 3,3 kPa (34 mbar). Porcje do badania – w przypadku syropu glukozowego lub syropu glukozowego w proszku – przygotowuje się, mieszając je przed suszeniem z wodą i ziemią okrzemkową.
2. Odczynnikiem używanym do oznaczania zawartości suchej masy metodą suszarki próżniowej jest oczyszczona ziemia okrzemkowa.
3. W celu otrzymania oczyszczonej ziemi okrzemkowej umieszcza się ją w lejku Buchnera i oczyszcza się przez kilkakrotne przemywanie rozcieńczonym kwasem solnym (1 ml stężonego kwasu, gęstość w 20 °C d = 1,19 g/ml na 1000 ml wody) aż do uzyskania odczynu kwaśnego popłuczyn (filtrowanej wody), to jest gdy pH osiągnie wartość większą niż 4. Następnie suszy się w suszarce w temperaturze 103 ą 2 °C i przechowuje w hermetycznie zamkniętym pojemniku.
4. Aparaturę służącą do oznaczania zawartości suchej masy stanowią:
1) suszarka próżniowa, zabezpieczona przed wyciekami, z termoregulacją, wyposażona w termometr i manometr próżniowy; suszarka powinna mieć budowę umożliwiającą szybkie przenoszenie ciepła do naczynek wagowych umieszczonych na półkach;
2) układ suszenia powietrznego składający się ze szklanego pojemnika wypełnionego świeżym, aktywowanym żelem krzemionkowym lub równorzędnym środkiem osuszającym wraz ze wskaźnikiem zawartości wody; pojemnik należy połączyć szeregowo z płuczką gazu zawierającą stężony kwas siarkowy i podłączoną do wlotu powietrza w suszarce;
3) pompa próżniowa umożliwiająca utrzymywanie w suszarce ciśnienia o wartości 3,3 kPa (34 mbar) lub mniejszego;
4) naczyńko do ważenia, z płaskim dnem, odporne na niszczące działanie próbek i warunki badawcze, o średnicy minimum 100 mm i wysokości minimum 300 mm;
5) bagietka szklana o długości większej niż naczyńko wagowe;
6) eksykator zawierający świeży, wysuszony żel krzemionkowy lub równorzędny środek osuszający wraz ze wskaźnikiem zawartości wody;
7) waga analityczna o dokładności 0,1 mg.
5. Przygotowanie próbki
Odważa się 30 g ziemi okrzemkowej, umieszcza się w naczyńku wagowym wyposażonym w szklaną bagietkę i całość umieszcza się w suszarce w temperaturze 70 ą 1 °C, zmniejszając ciśnienie do wartości 3,3 kPa (34 mbar) lub niższej.
Suszy się przez co najmniej 5 godzin, doprowadzając powolny strumień powietrza do suszarki przez układ suszenia – od czasu do czasu sprawdzając ciśnienie – i w miarę potrzeby korygując je.
Następnie przywraca się w suszarce ciśnienie atmosferyczne przez ostrożne zwiększenie poboru suchego powietrza; natychmiast umieszcza się naczyńko wagowe wraz ze szklaną bagietką w eksykatorze, studzi się i waży.
Do zlewki o pojemności 100 ml odważa się (z dokładnością do 1 mg) około 10 g próbki do analizy, dodaje się 10 ml ciepłej wody i przenosi się roztwór porcjami do naczyńka wagowego, używając do tego szklanej bagietki.
6. Suszenie próbki
Naczyńko wagowe zawierające badaną próbkę i szklaną bagietkę umieszcza się w suszarce, zmniejszając ciśnienie do 3,3 kPa (34 mbar) lub do niższego ciśnienia, i suszy się w temperaturze 70 ą 1 °C mniej więcej przez 20 godzin, pozwalając na powolny przepływ strumienia suchego powietrza przez suszarkę.
Następnie przywraca się w suszarce ciśnienie atmosferyczne, zwiększając ostrożnie dopływ suchego powietrza, natychmiast umieszcza się naczyńko wagowe wraz z wysuszoną próbką w eksykatorze, pozostawia się do ostudzenia i waży się z dokładnością do 1 mg.
Ponownie suszy się naczyńko z próbką przez 4 godziny, przywraca się w suszarce ciśnienie atmosferyczne, natychmiast umieszcza się naczyńko wraz z zawartością w eksykatorze, następnie pozostawia się do ostudzenia i waży.
7. Oznaczenie zawartości suchej masy
Określa się, czy ubytek masy osiągnął wartość stałą. Ubytek masy osiągnął wartość stałą, jeżeli różnica między dwoma ważeniami tego samego naczyńka nie przekracza 2 mg. Jeżeli różnica jest większa, ponownie suszy się naczyńko z próbką przez 4 godziny, następnie przywraca się w suszarce ciśnienie atmosferyczne, natychmiast umieszcza się naczyńko wraz z zawartością w eksykatorze, pozostawia się do ostudzenia i waży.
Do oznaczania suchej masy w próbkach dekstrozy bezwodnej lub dekstrozy jednowodnej nie stosuje się ziemi okrzemkowej i wody.
8. Wyrażanie wyników
Wyniki wyrażane są w procentach zawartości suchej masy, z uwzględnieniem powtarzalności wyników.
Zawartość suchej masy (D), wyrażoną w procentach, oblicza się według wzoru:
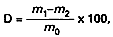
gdzie:
m0 – oznacza masę pobranej próbki przed suszeniem, w gramach,
m1 – oznacza masę naczynka do ważenia z ziemią okrzemkową, szklaną bagietką i badaną próbką po suszeniu, w gramach,
m2 – oznacza masę naczynka do ważenia z ziemią okrzemkową i szklaną bagietką, w gramach.
Różnica między wynikami dwóch oznaczeń przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 0,12 g na 100 g próbki.
Załącznik nr 9
METODA OZNACZANIA ZAWARTOŚCI POPIOŁU SIARCZANOWEGO W SYROPIE GLUKOZOWYM, SYROPIE GLUKOZOWYM W PROSZKU, W JEDNOWODNEJ GLUKOZIE (JEDNOWODNEJ DEKSTROZIE) I W BEZWODNEJ GLUKOZIE (BEZWODNEJ DEKSTROZIE)
1. Metoda oznaczania zawartości popiołu siarczanowego w syropie polega na oznaczeniu masy badanej próbki po spopieleniu w atmosferze utleniającej w temperaturze 525 °C, w obecności kwasu siarkowego, a następnie na obliczeniu jej procentowej zawartości (m/m).
2. Odczynnikiem używanym do oznaczania zawartości popiołu siarczanowego jest rozcieńczony roztwór kwasu siarkowego.
3. W celu otrzymania rozcieńczonego roztworu kwasu siarkowego powoli i ostrożnie dodaje się 100 ml stężonego kwasu siarkowego (gęstość w temperaturze 20 °C d = 1,84 g/ml; 96% m/m) do 300 ml wody.
4. Sprzęt laboratoryjny służący do oznaczania popiołu siarczanowego stanowią:
1) elektryczny piec muflowy, wyposażony w termometr, działający w temperaturze 525 ą 25 °C;
2) waga analityczna, o dokładności 0,1 mg;
3) tygle do spopielania: platynowe lub kwarcowe, o odpowiedniej pojemności;
4) eksykator, zawierający świeży żel krzemionkowy lub równorzędny środek osuszający, wyposażony we wskaźnik zawartości wody.
5. Przygotowanie próbki
Wypraża się tygiel w temperaturze spopielania (525 °C), studzi się w eksykatorze i waży. Następnie odważa się do niego, z dokładnością do 0,1 mg, 5 g syropu glukozowego lub syropu glukozowego w proszku lub około 10 g dekstrozy jednowodnej lub dekstrozy bezwodnej i dodaje się 5 ml roztworu kwasu siarkowego.
6. Wykonanie oznaczenia
W celu zwęglenia próbki ostrożnie podgrzewa się próbkę w tyglu nad płomieniem lub na płycie grzejnej aż do całkowitego jej zwęglenia. Proces zwęglania, podczas którego wydzielają się pary z próbki ulegającej spaleniu, przeprowadza się pod wyciągiem laboratoryjnym.
Po zwęgleniu próbki umieszcza się tygiel w piecu muflowym podgrzanym do temperatury 525 ą 25 °C aż do czasu uzyskania białego popiołu. Czas trwania wynosi około 2 godzin.
Pozostawia się tygiel z próbką przez 30 minut aż do ostudzenia w eksykatorze i waży się.
7. Wyrażanie wyników
Wyniki są wyrażane w procentach masy analizowanej próbki, z uwzględnieniem powtarzalności wyników.
Zawartość popiołu siarczanowego wyrażoną w procentach masy analizowanej próbki oblicza się według wzoru:
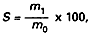
gdzie:
m1 – oznacza masę popiołu, w gramach,
m0 – oznacza masę badanej próbki, w gramach.
Przyjmuje się, że różnica między wynikami dwóch oznaczeń przeprowadzonych równocześnie lub w krótkim odstępie czasu z tą samą próbką, przez tego samego analityka i w tych samych warunkach, nie powinna przekraczać 2% ich średniej arytmetycznej.
8. Uwagi
W celu zapobieżenia nadmiernemu tworzeniu piany kwas siarkowy dodaje się w niewielkich ilościach.
Podczas pierwszego zwęglania podejmuje się wszelkie niezbędne środki w celu zapobieżenia stracie próbki lub popiołu poprzez nadmierne spęcznienie próbki.
Jeżeli trudno jest całkowicie spopielić próbkę (pozostałość zawiera czarne cząsteczki), należy tygiel wyjąć z pieca muflowego, a pozostałość po ostudzeniu zwilżyć kilkoma kroplami wody i umieścić ponownie w piecu.
- Data ogłoszenia: 2004-03-09
- Data wejścia w życie: 2004-05-01
- Data obowiązywania: 2004-05-01

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA