REKLAMA
Dziennik Ustaw - rok 2003 nr 138 poz. 1318
ROZPORZĄDZENIE MINISTRA ROLNICTWA I ROZWOJU WSI1)
z dnia 3 lipca 2003 r
w sprawie metod analiz alkoholu etylowego rolniczego oraz metod pobierania próbek do celów urzędowej kontroli pod względem jakości handlowej
Na podstawie art. 20 ustawy z dnia 13 września 2002 r. o napojach spirytusowych (Dz. U. Nr 166, poz. 1362) zarządza się, co następuje:
1) oznaczania zawartości alkoholu etylowego rolniczego wyrażonego w procentach objętościowych – wykonywanego metodą, która jest określona w załączniku nr 1 do rozporządzenia;
2) oceny barwy lub klarowności – wykonywanej metodą, która jest określona w załączniku nr 2 do rozporządzenia;
3) pomiaru czasu odbarwiania roztworu nadmanganianu(VII)potasu (próba Langa) – wykonywanego metodą, która jest określona w załączniku nr 3 do rozporządzenia;
4) oznaczania zawartości aldehydów – wykonywanego metodą, która jest określona w załączniku nr 4 do rozporządzenia;
5) oznaczania kwasowości całkowitej – wykonywanego metodą, która jest określona w załączniku nr 5 do rozporządzenia;
6) oznaczania zawartości estrów – wykonywanego metodą, która jest określona w załączniku nr 6 do rozporządzenia,
7) oznaczania zawartości lotnych zasad azotowych – wykonywanego metodą, która jest określona w załączniku nr 7 do rozporządzenia;
8) oznaczania zawartości suchej pozostałości po odparowaniu – wykonywanego metodą, która jest określona w załączniku nr 8 do rozporządzenia;
9) próby na obecność furfuralu – wykonywanej metodą, która jest określona w załączniku nr 9 do rozporządzenia;
10) testu UV – wykonywanego metodą, która jest określona w załączniku nr 10 do rozporządzenia;
11) oznaczania zawartości izotopu węgla 14C – wykonywanego metodą, która jest określona w załączniku nr 11 do rozporządzenia.
2. W przypadku, o którym mowa w ust. 1, za ostateczny uznaje się wynik jednej z metod wymienionych w § 2.
Minister Rolnictwa i Rozwoju Wsi: W. Olejniczak
|
|
1) Minister Rolnictwa i Rozwoju Wsi kieruje działem administracji rządowej – rynki rolne, na podstawie § 1 ust 2 pkt 3 rozporządzenia Prezesa Rady Ministrów z dnia 29 marca 2002 r. w sprawie szczegółowego zakresu działania Ministra Rolnictwa Rozwoju Wsi (Dz. U. Nr 32, poz. 305).
Załączniki do rozporządzenia Ministra Rolnictwa i Rozwoju Wsi
z dnia 3 lipca 2003 r. (poz. 1318)
Załącznik nr 1
OZNACZANIE ZAWARTOŚCI ALKOHOLU ETYLOWEGO ROLNICZEGO WYRAŻONEJ W PROCENTACH OBJĘTOŚCIOWYCH
Oznaczenie zawartości alkoholu etylowego rolniczego w procentach objętościowych wykonuje się jedną z następujących metod:
1) przy użyciu alkoholomierza lub termoalkoholomierza;
2) przy użyciu piknometru;
3) przy użyciu elektronicznego analizatora gęstości.
Przygotowanie próbki do analizy
Pobrana próbka alkoholu etylowego rolniczego jest przechowywana w hermetycznym pojemniku zabezpieczającym ją przed dostępem powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
l. Oznaczanie zawartości alkoholu etylowego rolniczego przy użyciu alkoholomierza lub termoalkoholomierza
Oznaczanie zawartości alkoholu etylowego rolniczego w procentach objętościowych polega na ustaleniu wskazań alkoholomierza i termometru, a następnie na odczytaniu odpowiadającej im zawartości alkoholu etylowego w Tablicach Alkoholometrycznych określonych w przepisach prawa o miarach.
1. APARATURA
Do oznaczania zawartości alkoholu stosuje się:
1) cylinder szklany przezroczysty;
2) alkoholomierz lub termoalkoholomierz z podziałką elementarną 0,1; 0,2; 0,5; 1% obj., mający cechy legalizacyjne;
3) termometr z podziałką elementarną 0,50 C lub 10 C, mający cechy legalizacyjne.
2. WYKONANIE OZNACZANIA
Do cylindra wlewa się taką ilość próbki, aby poziom cieczy znajdował się kilka centymetrów poniżej brzegu cylindra, a zanurzenie alkoholomierza nie spowodowało wylania cieczy. W cylindrze z badaną próbką powoli zanurza się czysty i suchy alkoholomierz tak, aby nie dotykał ścianek i dna cylindra. Wskazania alkoholomierza odczytuje się po ustaleniu temperatury próbki, według dolnej linii menisku. Podczas analizy odczytuje się temperaturę próbki. Na podstawie wskazań alkoholomierza i termometru zawartość alkoholu etylowego w procentach objętościowych odczytuje się z Tablic Alkoholometrycznych.
3. OBLICZANIE WYNIKU
Zawartość alkoholu etylowego w % objętościowych odczytuje się z Tablic Alkoholometrycznych. Wynikiem oznaczania zawartości alkoholu etylowego jest średnia arytmetyczna uzyskana z co najmniej dwóch równolegle wykonanych pomiarów, różniących się między sobą nie więcej niż o dokładność odczytu z alkoholomierza.
II. Oznaczanie zawartości alkoholu etylowego rolniczego przy użyciu piknometru
Oznaczanie zawartości alkoholu etylowego przy użyciu piknometru polega na oznaczaniu gęstości badanej próbki w temperaturze 200 C i odczytaniu odpowiadającej jej zawartości alkoholu etylowego z Tablic Alkoholometrycznych.
1. APARATURA
Do oznaczania zawartości alkoholu stosuje się:
1) wagę analityczną o dokładności 0,0001 g;
2) termostat lub łaźnię wodną;
3) piknometr o pojemności 50 ml.
2. WYKONANIE OZNACZANIA
1) czysty i suchy piknometr waży się z dokładnością do 0,0002 g, napełnia się wodą destylowaną powyżej kreski i wstawia do łaźni wodnej o temperaturze 20 ± 10 C na 30 minut. Wyrównuje się poziom wody do kreski, stosując rurkę kapilarną lub rulonik z bibuły. Piknometr po wytarciu do sucha waży się;
2) czynności określone w pkt 1 powtarza się, biorąc zamiast wody badaną próbkę.
3. OBLICZANIE WYNIKU
Gęstość badanej próbki (ρ20) w temperaturze 200 C, wyrażoną w g/ml oblicza się według wzoru:
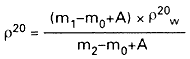
gdzie:
m0 – oznacza masę pustego piknometru w gramach,
m1 – oznacza masę piknometru z badaną próbką w gramach,
m2 – oznacza masę piknometru z wodą w gramach,
A – oznacza poprawkę na ważenie w powietrzu, która wynosi 0,0012 x (m2-m0), gdzie 0,0012 jest gęstością powietrza.
ρ20w – oznacza gęstość wody w temperaturze 200 C, która wynosi 0,9982 g/ml.
Zawartość alkoholu etylowego w % objętościowych odpowiadającą obliczonej gęstości próbki odczytuje się z Tablic Alkoholometrycznych. Wynikiem oznaczania zawartości alkoholu etylowego jest średnia arytmetyczna uzyskana z co najmniej dwóch równolegle wykonanych pomiarów różniących się pomiędzy sobą nie więcej niż o 0,1% obj.
III. Oznaczanie zawartości alkoholu etylowego przy użyciu elektronicznego analizatora gęstości
1. APARATURA
Do oznaczania zawartości alkoholu stosuje się elektroniczny analizator gęstości o dokładności pomiaru co najmniej 0,08%, wyskalowany w procentach objętościowych.
2. WYKONANIE OZNACZANIA
Pomiaru dokonuje się przy użyciu elektronicznego analizatora gęstości w temperaturze 200 C, zgodnie z instrukcją obsługi. Odczytuje się zawartość alkoholu etylowego w procentach objętościowych.
3. OBLICZANIE WYNIKU
Wynikiem oznaczania zawartości alkoholu etylowego jest średnia arytmetyczna uzyskana co najmniej z dwóch równolegle wykonanych pomiarów, różniących się między sobą nie więcej niż o 0,08% obj.
Załącznik nr 2
OCENA BARWY LUB KLAROWNOŚCI
Barwa i klarowność są oceniane wzrokowo przez porównanie próbki z wodą destylowaną na białym i czarnym tle.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Pobrana próbka alkoholu etylowego rolniczego jest przechowywana w hermetycznym pojemniku zabezpieczającym ją przed dostępem powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
2. APARATURA
Do wykonania oceny barwy lub klarowności stosuje się cylindry wykonane z bezbarwnego szkła o wysokości co najmniej 40 cm.
3. WYKONANIE OZNACZANIA
Dwa szklane cylindry umieszcza się obok siebie na białym lub na czarnym tle. Następnie jeden cylinder napełnia się próbką do wysokości około 40 cm, a drugi – wodą do tej samej wysokości. Patrząc z góry na oba cylindry porównuje się barwę i klarowność próbki alkoholu etylowego rolniczego z barwą i klarownością wody.
4. OBLICZANIE WYNIKU
Badana próbka alkoholu etylowego rolniczego jest bezbarwna, gdy jej barwa jest identyczna z barwą wody destylowanej.
Badana próbka alkoholu etylowego rolniczego jest klarowna, jeżeli światło przechodzące przez próbkę nie ulega rozproszeniu, powodując jej opalizację.
Załącznik nr 3
POMIAR CZASU ODBARWIANIA ROZTWORU NADMANGANIANU(VII)POTASU (próba Langa)
Czas odbarwiania roztworu nadmanganianu(VII)potasu jest to czas liczony w minutach, potrzebny do zrównania barwy próbki z barwą wzorca, po dodaniu roztworu nadmanganianu(VII)potasu.
1. PRZYGOTOWANIE DO ANALIZY
Pobrana próbka alkoholu etylowego rolniczego jest przechowywana w hermetycznym pojemniku zabezpieczającym ją przed dostępem powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
2. ODCZYNNIKI
Do oznaczania stosuje się następujące odczynniki chemiczne o czystości analitycznej:
1) roztwór nadmanganianu(VII)potasu o stężeniu 1 mmol/l, przygotowany bezpośrednio przed użyciem;
2) roztwór barwny A (czerwony), przygotowany w następujący sposób: w kolbie miarowej o pojemności 1000 ml rozpuszcza się 59,50 g chlorku kobaltu(ll) (CoCI2 ۰ 6H2O) w roztworze kwasu chlorowodorowego sporządzonego z 25 ml kwasu chlorowodorowego o gęstości ρ = 1,19 g/ml i 975 ml wody, a następnie uzupełnia się do objętości 1000 ml roztworem kwasu;
3) roztwór barwny B (żółty), przygotowany w następujący sposób: w kolbie miarowej o pojemności 1000 ml rozpuszcza się 45,00 g chlorku żelaza(lll) (FeCI3 ۰ 6H2O) w roztworze kwasu chlorowodorowego sporządzonego z 25 ml kwasu chlorowodorowego o gęstości ρ = 1,19 g/ml i 975 ml wody, a następnie uzupełnia się do objętości 1000 ml roztworem kwasu;
4) wzorcowy roztwór barwny, przygotowany w następujący sposób: do kolby miarowej o pojemności 100 ml odmierza się pipetą 13 ml roztworu A i 5,5 ml roztworu B, po czym uzupełnia do objętości 100 ml wodą o temperaturze 200 C.
Roztwory barwne A i B można przechowywać w temperaturze 40 C kilka miesięcy, natomiast wzorcowy roztwór barwny przygotowuje się bezpośrednio przed użyciem.
3. APARATURA
Do oznaczania stosuje się:
1) probówki Nesslera o pojemności 100 ml wykonane z przezroczystego, bezbarwnego szkła, z podziałką do 50 ml i korkiem z matowego szkła, lub probówki zwykłe, bezbarwne o średnicy około 20 mm;
2) pipety o pojemności: 1, 2, 5, 10 i 50 ml;
3) termometr o zakresie pomiarowym do 500 C z działką elementarną 0,10 C lub 0,20 C;
4) wagę analityczną;
5) łaźnię wodną z możliwością utrzymania temperatury 20 ± 0,50 C;
6) kolby miarowe o pojemności 100 i 1000 ml z doszlifowanymi korkami.
4. WYKONANIE OZNACZANIA
Do probówki Nesslera odmierza się 50 ml z próbki (w przypadku użycia zwykłej probówki odmierza się 10 ml). Probówki umieszcza się w łaźni wodnej o temperaturze 200 C. Następnie do probówki dodaje się 1 ml lub 5 ml (w zależności od ilości użytej próbki) roztworu nadmanganianu(VII)potasu, miesza się, odnotowuje czas i pozostawia w łaźni wodnej w temperaturze 200 C. Do drugiej probówki Nesslera odmierza się 50 ml wzorcowego roztworu barwnego, a w przypadku stosowania zwykłej probówki o takiej samej średnicy 10 ml wzorcowego roztworu barwnego. Obserwuje się zmianę barwy próbki i porównuje się ją z barwą wzorca na białym tle. Odnotowuje się czas, w jakim próbka osiągnęła tę samą barwę co roztwór wzorcowy.
5. OBLICZANIE WYNIKU
Jako wynik końcowy pomiaru podaje się liczony w minutach czas zrównania barwy badanej próbki z barwą wzorca.
Dla alkoholu etylowego rolniczego czas ten wynosi co najmniej 18 minut przy temperaturze 200 C.
6. POWTARZALNOŚĆ
Różnica czasu między dwoma oznaczeniami przeprowadzonymi jednocześnie lub w krótkim odstępie czasu przez tego samego analityka, na tej samej próbce i w takich samych warunkach, nie powinna przekraczać dwóch minut.
Załącznik nr 4
OZNACZANIE ZAWARTOŚCI ALDEHYDÓW
Zawartość aldehydów jest wyrażana jako aldehyd octowy.
Oznaczanie zawartości aldehydów polega na reakcji aldehydów z odczynnikiem Schiff΄a i porównaniu intensywności zabarwienia związków kompleksowych powstałych w badanej próbce z zabarwieniem roztworów wzorcowych o znanej zawartości aldehydu octowego.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Próbka do analizy jest przechowywana w czystej kolbie zamkniętej doszlifowanym korkiem przemytym alkoholem lub korkiem zawiniętym w folię cynową lub aluminiową w warunkach zapewniających brak dostępu powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
2. ODCZYNNIKI
Do oznaczania zawartości aldehydów stosuje się następujące odczynniki chemiczne o czystości analitycznej:
1) chlorowodorek p-rosaniliny (fuksyna zasadowa);
2) siarczan (IV) sodu lub bezwodny wodorosiarczan (IV)sodu;
3) kwas chlorowodorowy, gęstość ρ20 = 1,19 g/ml;
4) sproszkowany węgiel aktywny;
5) roztwór skrobi, przygotowany w następujący sposób: 1 g skrobi miesza się z 5 mg jodku rtęci (Hgl2 – środek konserwujący), następnie dodaje się niewielką ilość zimnej wody, miesza się i przenosi do kolby zawierającej 500 ml wrzącej wody; całość miesza się i gotuje przez 5 minut, po czym studzi się i przefiltrowuje;
6) 1-amino-etanol CH3CH(NH2)OH, oczyszczony i wysuszony, przygotowany w następujący sposób: 5 g 1-amino-etanolu rozpuszcza się w około 15 ml alkoholu etylowego 100% obj.; następnie dodaje się około 50 ml suchego eteru etylowego i pozostawia na kilka godzin w lodówce; następnie odfiltrowuje się kryształy i przepłukuje suchym eterem etylowym; uzyskany oczyszczony 1-amino-etanol suszy się przez 3–4godziny w eksykatorze pod zmniejszonym ciśnieniem nad kwasem siarkowym (VI). Jeżeli oczyszczony 1-amino-etanol nie ma białego koloru, należy powtórzyć proces rekrystalizacji;
7) roztwór jodu o stężeniu 0,05 mol/l;
8) odczynnik Schiff΄a przygotowany w następujący sposób: w kolbie miarowej o pojemności 2000 ml rozpuszcza się 5,0 g sproszkowanego chlorowodorku p-rosaniliny w 1000 ml gorącej wody i pozostawia się w łaźni wodnej do całkowitego rozpuszczenia. W ok. 200 ml wody rozpuszcza się 30 g siarczanu (IV) sodu (lub równoważną ilość wodorosiarczanu (IV sodu) i dodaje do schłodzonego roztworu p-rosaniliny, i pozostawia na 10 minut; następnie dodaje się 60 ml kwasu chlorowodorowego; jeżeli roztwór jest bezbarwny lub wykazuje lekkie brązowe zabarwienie, uzupełnia się wodą do objętości 2000 ml; w przypadku gdy roztwór nie jest bezbarwny, przefiltrowuje się go z odrobiną aktywnego węgla na złożonym filtrze, a następnie uzupełnia wodą do objętości 2000 ml.
Dopuszcza się inne metody przygotowania odczynnika Schiff΄a, pod warunkiem że podczas testu kontrolnego nie daje on reakcji barwnej z alkoholem etylowym bezaldehydowym i daje widoczne różowe zabarwienie z roztworem wzorcowym o zawartości 0,1 g aldehydu octowego w 1 hl spirytusu 100% obj.
Odczynnik Schiff΄a powinien być przygotowany co najmniej 14 dni przed użyciem. Zawartość wolnego SO2 w odczynniku powinna wynosić od 2,8 do 6,0 mmol w 100 ml, a pH odczynnika powinno wynosić 1.
Oznaczanie wolnego SO2:
Do kolby stożkowej o pojemności 250 ml odmierza się 10 ml odczynnika Schiff΄a i dodaje się 200 ml wody i 5 ml roztworu skrobi, a następnie miareczkuje się roztworem jodu.
Ilość wolnego SO2 w odczynniku w milimolach wolnego SO2 w 100 ml odczynnika oblicza się, dzieląc przez dwa zużytą ilość mililitrów roztworu jodu.
Jeśli zawartość wolnego SO2 jest mniejsza lub większa od wskazanego zakresu, należy podwyższyć ją przez dodanie obliczonej ilości pirosiarczynu sodowego (0,126 g Na2SO3/100 ml odczynnika na każdy brakujący mmol SO2) lub obniżyć poprzez przepuszczanie przez odczynnik powietrza.
3. APARATURA
Do oznaczania zawartości aldehydów stosuje się:
1) probówki kolorymetryczne o pojemności 20 ml zaopatrzone w korek z matowego szkła;
2) pipety o pojemności: 1, 2, 3, 5, 10 ml;
3) łaźnię wodną z możliwością utrzymania temperatury 20 ± 0,50 C;
4) spektrofotometr UV-VIS, umożliwiający oznaczanie absorbancji roztworów przy długości fali λ = 546 nm z kuwetami o długości drogi optycznej 50 mm.
4. WYKONANIE OZNACZANIA
Zawartość alkoholu etylowego rolniczego w próbce powinna wynosić co najmniej 90% obj. Jeżeli jest go mniej, zawartość tę zwiększa się przez dodanie odpowiedniej ilości alkoholu etylowego bezaldehydowego.
Przygotowanie roboczych roztworów wzorcowych
W kolbie miarowej o pojemności 1 000 ml rozpuszcza się 1,3860 g (odważonego na wadze analitycznej) oczyszczonego i osuszonego 1-amino-etanolu w alkoholu etylowym bezaldehydowym i uzupełnia do objętości 1 000 ml. Przygotowany w ten sposób 1 litr roztworu wzorcowego zawiera 1 g aldehydu octowego.
Następnie przygotowuje się poprzez odpowiednie rozcieńczenie roztworu wzorcowego (w dwóch seriach) po 10 roztworów wzorcowych zawierających od 0,1 do 1,0 mg aldehydu octowego w 100 ml roztworu.
Wyznaczanie krzywej wzorcowej dla roboczych roztworów wzorcowych
Do probówek kolorymetrycznych odmierza się pipetą po 5 ml roboczych roztworów wzorcowych; w tym samym czasie do jednej z probówek odmierza się zamiast roboczego roztworu wzorcowego 5 ml alkoholu etylowego bezaldehydowego (96% obj.). Następnie do każdej probówki dodaje się po 5 ml wody, zawartość probówek miesza się i umieszcza w łaźni wodnej o temperaturze 200 C. Po ustaleniu się temperatury w probówkach dodaje się 5 ml odczynnika Schiff΄a. Probówki zamyka się korkiem, miesza i pozostawia w łaźni wodnej. Po 20 minutach od dodania odczynnika Schiff΄a zawartość poszczególnych probówek przelewa się do kuwet spektrofotometrycznych i oznacza się wartość absorbancji roztworów przy długości fali λ = 546 nm.
Następnie wykreśla się krzywą wzorcową dla roboczych roztworów wzorcowych aldehydu octowego w układzie A = f(c), gdzie: A – oznacza wartość absorbancji, c – stężenie aldehydu octowego w roboczym roztworze wzorcowym w g/hl.
Oznaczanie zawartości aldehydów w próbce
Do probówki kolorymetrycznej odmierza się pipetą 5 ml z badanej próbki. Następnie dodaje się 5 ml wody, zawartość probówki miesza się i umieszcza w łaźni wodnej o temperaturze 200 C. Po ustaleniu temperatury w próbówce, dodaje się 5 ml odczynnika Schiff΄a. Probówkę zamyka się korkiem, miesza i pozostawia w łaźni wodnej. Po 20 minutach od dodania odczynnika Schiff΄a zawartość probówki przelewa się do kuwety spektrofotometrycznej i oznacza się wartość absorbancji roztworu przy długości fali λ = 546 nm. Następnie z krzywej wzorcowej odczytuje się dla zmierzonej wartości absorbancji stężenie aldehydu octowego w g/hl alkoholu etylowego 100% obj.
5. OBLICZANIE WYNIKU
Zawartość aldehydów w badanej (Az) próbce, w g/hl alkoholu etylowego 100% obj., wyrażoną jako aldehyd octowy, oblicza się według wzoru:
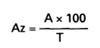
gdzie:
A – oznacza zawartość aldehydu octowego w próbce, odczytaną z krzywej wzorcowej w g/hl,
T – oznacza zawartość alkoholu etylowego w próbce w procentach objętościowych.
6. POWTARZALNOŚĆ
Różnica między wynikami dwóch oznaczeń przeprowadzonych jednocześnie lub w krótkim odstępie czasu, przez tego samego analityka, na tej samej próbce, w takich samych warunkach nie powinna przekraczać 0,1 g aldehydu na hektolitry alkoholu etylowego 100% obj.
Załącznik nr 5
OZNACZANIE KWASOWOŚCI CAŁKOWITEJ
Kwasowość całkowita jest wyrażana jako kwas octowy.
Metoda oznaczania kwasowości całkowitej polega na zmiareczkowaniu mianowanym roztworem wodorotlenku sodu odgazowanej próbki wobec indygo, karminu i czerwieni fenylowej jako wskaźników.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Próbka do analizy jest przechowywana w czystej kolbie zamkniętej doszlifowanym korkiem przemytym alkoholem lub korkiem zawiniętym w folię cynową lub aluminiową, w warunkach zapewniających brak dostępu powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
2. ODCZYNNIKI
Do oznaczania kwasowości całkowitej stosuje się następujące odczynniki chemiczne o czystości analitycznej:
1) roztwory wodorotlenku sodowego 0,01 mol/l i 0,1 mol/l, przechowywane w sposób ograniczający do minimum kontakt z dwutlenkiem węgla;
2) roztwór indygokarminu (A), przygotowany w następujący sposób: 0,2 g indygokarminu rozpuszcza się w 40 ml wody i dopełnia alkoholem etylowym do 100 g;
3) roztwór czerwieni fenolowej (B), przygotowany w następujący sposób: 0,2 g czerwieni fenolowej rozpuszcza się w 6 ml roztworu wodorotlenku sodu 0,1 mol/l i dopełnia wodą do objętości 100 ml w kolbie miarowej.
3. APARATURA
Do oznaczania kwasowości całkowitej stosuje się:
1) biuretę lub automatyczne urządzenie do miareczkowania;
2) pipetę o pojemności 100 ml;
3) kolbę kulistą o pojemności 250 ml z doszlifowanym korkiem z matowego szkła;
4) chłodnicę zwrotną z doszlifowanym korkiem z matowego szkła.
4. WYKONANIE OZNACZANIA
Do kolby kulistej odmierza się pipetą 100 ml z próbki, następnie dodaje się kilka kawałków porcelany. Kolbę z roztworem umieszcza się w łaźni wodnej i doprowadza do wrzenia pod chłodnicą zwrotną. Do gorącego roztworu dodaje się po jednej kropli roztworów wskaźnikowych A i B. Następnie miareczkuje się roztworem wodorotlenku sodowego 0,01 mol/l do momentu pojawienia się oznak zmiany barwy z zielono-żółtej na fioletową.
5. OBLICZANIE WYNIKU
Kwasowość ogólną (Ko) wyrażoną w postaci kwasu octowego, w g/hl alkoholu etylowego 100% obj., oblicza się według wzoru:
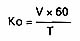
gdzie
V – oznacza ilość ml roztworu wodorotlenku sodowego 0,01 mol/l, zużytą podczas miareczkowania,
T – oznacza zawartość alkoholu etylowego w próbce, w procentach objętościowych.
6. POWTARZALNOŚĆ
Różnica między wynikami dwóch oznaczeń przeprowadzonych jednocześnie bądź w krótkim odstępie czasu, przez tego samego analityka, na tej samej próbce i w takich samych warunkach, nie powinna przekraczać 0,1 g/hl alkoholu etylowego 100% obj.
Załącznik nr 6
OZNACZANIE ZAWARTOŚCI ESTRÓW
Zawartość estrów jest wyrażana jako octan etylu.
Metoda oznaczania zawartości estrów polega na pomiarze absorbancji przy długości fali λ = 525 nm kompleksów barwnych otrzymanych w wyniku reakcji kwasów hydroksyloaminowych, powstałych podczas reakcji estrów z chlorowodorkiem hydroksyloaminy w roztworach alkalicznych z jonami żelazowymi w roztworach kwaśnych.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Próbka do analizy jest przechowywana w czystej kolbie zamkniętej doszlifowanym korkiem przemytym alkoholem lub korkiem zawiniętym w folię cynową lub aluminiową, w warunkach zapewniających brak dostępu powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbką miesza się w celu jej ujednolicenia.
2. ODCZYNNIKI
Do oznaczania zawartości estrów stosuje się następujące odczynniki chemiczne o czystości analitycznej:
1) kwas chlorowodorowy 4 mol/l;
2) roztwór chlorku żelaza (III) 0,37 mol/l w kwasie chlorowodorowym 1 mol/l;
3) chlorowodorek hydroksyloaminy 2 mol/l, przechowywany w lodowce;
4) roztwór wodorotlenku sodu 3,5 mol/l;
5) wzorcowe roztwory octanu etylu zawierające 0,0; 0,2; 0,4; 0,6; 0,8 i 1,0 g octanu etylu na hektolitr wolnego od estrów alkoholu etylowego 96% obj.
3. APARATURA
Do oznaczania zawartości estrów stosuje się:
1) spektrofotometr UV-VIS, umożliwiający oznaczanie absorbancji roztworów przy długości fali λ = 525 nm;
2) kuwety spektrofotometryczne o długości drogi optycznej 50 mm;
3) łaźnię wodną z możliwością utrzymania temperatury 20 ± 0,50 C;
4) probówki kolorymetryczne o pojemności 20 ml wykonane z matowego szkła z doszlifowanymi korkami.
4. WYKONANIE OZNACZANIA
Przygotowanie roboczych roztworów wzorcowych
Na wadze analitycznej odważa się 1,0 g octanu etylu. Odważony octan etylu wsypuje się do kolby miarowej o pojemności 1 000 ml zawierającej alkohol etylowy wolny od estrów i uzupełnia do objętości 1 000 ml. Przygotowany w ten sposób 1 litr roztworu wzorcowego zawiera 1 g octanu etylowego.
Następnie przygotowuje się, przez rozcieńczenie roztworu wzorcowego (w dwóch seriach) po 10 roboczych roztworów wzorcowych zawierających od 0,1 do 2,0 mg octanu etylu w 100 ml.
Wyznaczenie krzywej wzorcowej dla roboczych roztworów wzorcowych
Do probówek kolorymetrycznych odmierza się po 10 ml roboczych roztworów wzorcowych i dodaje 2 ml roztworu chlorowodorku hydroksyloaminy. Jednocześnie przygotowuje się próbę ślepą, używając zamiast roboczego roztworu wzorcowego alkoholu etylowego 96% obj. wolnego od estrów. Następnie do każdej probówki dodaje się po 2 ml wodorotlenku sodu, probówki zamyka się korkami i dokładnie wstrząsa. Probówki umieszcza się w łaźni wodnej o temperaturze 200 C. Po 15 minutach dodaje się do każdej probówki 2 ml kwasu chlorowodorowego i wstrząsa. Następnie dodaje się 2 ml roztworu chlorku żelazowego i dokładnie miesza.
Zawartość probówek przelewa się do kuwet spektrofotometrycznych i oznacza absorbancją przy długości fali λ = 525 nm, stosując ślepą próbę jako odnośnik.
Następnie wykreśla się krzywą wzorcową w układzie A = f(c), gdzie A – oznacza wartość absorbancji, c – stężenie estrów w roboczym roztworze wzorcowym w g/hl.
Oznaczanie zawartości estrów w badanej próbce
Do probówki kolometrycznej odmierza się po 10 ml z badanej próbki i dodaje 2 ml roztworu wodorochlorku hydroksyloaminy. Następnie do probówki dodaje się 2 ml wodorotlenku sodu, probówkę zamyka się korkiem i wstrząsa. Probówkę umieszcza się w łaźni wodnej o temperaturze 200 C. Po 15 minutach dodaje się 2 ml kwasu chlorowodorowego i wstrząsa się. Następnie dodaje się 2 ml roztworu chlorku żelazowego i dokładnie miesza.
Zawartość probówki przelewa się do kuwety spektrofotometrycznej i oznacza absorbancję przy długości fali λ = 525 nm.
5. OBLICZANIE WYNIKU
Zawartość estrów (E), w g/hl w przeliczeniu na octan etylu oblicza się według wzoru:
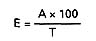
gdzie A – oznacza zawartość estrów odczytaną z krzywej wzorcowej w g/hl,
T – oznacza zawartość alkoholu etylowego w próbce w procentach objętościowych.
6. POWTARZALNOŚĆ
Różnica między wynikami dwóch oznaczeń, przeprowadzonych jednocześnie lub w krótkim odstępie czasu przez tego samego analityka, na tej samej próbce i w takich samych warunkach, nie powinna przekraczać 0,1 g estrów wyrażanej jako octan etylu, na hektolitry alkoholu etylowego 100% obj.
Załącznik nr 7
OZNACZANIE ZAWARTOŚCI LOTNYCH ZASAD AZOTOWYCH
Lotne zasady azotowe są wyrażane jako azot w alkoholu etylowym rolniczym. Metoda oznaczania lotnych zasad azotowych polega na uwolnieniu związków azotowych mikrodyfuzyjną techniką Conway'a. Uwolnione związki azotowe w postaci amoniaku miareczkuje się mianowanym roztworem kwasu chlorowodorowego.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Próbka do analizy jest przechowywana w czystej kolbie zamkniętej doszlifowanym korkiem przemytym alkoholem lub korkiem zawiniętym w folię cynową lub aluminiową, w warunkach zapewniających brak dostępu powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
2. ODCZYNNIKI
Do oznaczania zawartości lotnych zasad azotowych stosuje się następujące odczynniki chemiczne o czystości analitycznej:
1) kwas siarkowy (VI) 1 mol/l;
2) roztwór wskaźnikowy kwasu bornego, przygotowany w następujący sposób: 10 g kwasu bornego, 8 mg zieleni bromokrezolowej oraz 4 mg czerwieni metylowej rozpuszcza się w 30% obj. propan-2-olu oraz dopełnia 30% objętościowym propan-2-olem do objętości 1 000 ml;
3) roztwór wodorotlenku potasu 500 g/l wolny od dwutlenku węgla;
4) kwas chlorowodorowy 0,02 mol/l.
3. APARATURA
Do oznaczania zawartości lotnych zasad azotowych stosuje się:
1) parownicę o pojemności umożliwiającej umieszczenie w niej 50 ml próbki;
2) łaźnię wodną;
3) mikrobiuretę o pojemności od 2 do 5 ml, z podziałką co 0,01 ml;
4) naczynie Conway'a ze ściśle dopasowaną pokrywą; opis i zalecane wymiary są przedstawione na schemacie.
Schemat naczynia Conway'a
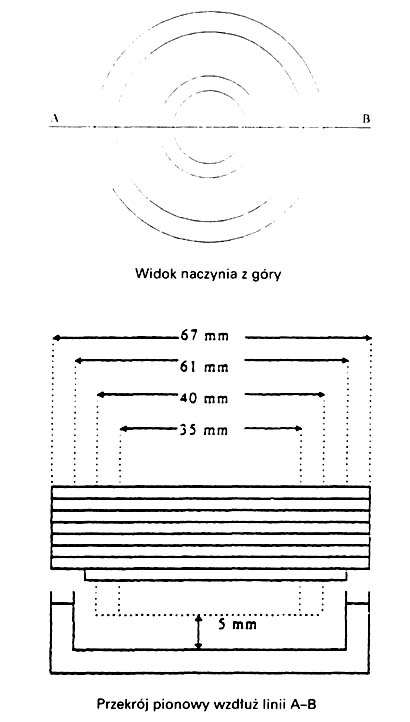
4. WYKONANIE OZNACZANIA
Do szklanej parownicy odmierza się pipetą 50 ml z próbki (o zawartości azotu ocenianej na mniejszą niż 0,2 g/hl próbki – odmierza się 200 ml próbki), następnie dodaje się 1 ml kwasu siarkowego (VI), umieszcza parownicę w łaźni wodnej i odparowuje tę próbkę do momentu, gdy pozostanie z niej około 1 ml.
Do wewnętrznej komory naczynia Conway'a odmierza się pipetą 1 ml wskaźnikowego roztworu kwasu bornego. Następnie zawartość parownicy przenosi się ilościowo (popłukując niewielką ilością wody) na jedną stronę zewnętrznej komory naczynia Conway'a, zaś na drugą stronę zewnętrznej komory naczynia Conway'a odmierza się 1 ml roztworu wodorotlenku potasowego, a następnie niezwłocznie po nalaniu naczynie Conway'a nakrywa się szczelnie.
Oba roztwory znajdujące się w komorze zewnętrznej miesza się, uważając, aby płyn nie przeciekał z jednej komory do drugiej, następnie pozostawia się na dwie godziny. Wydzielony w komorze wewnętrznej amoniak miareczkuje się przy pomocy roztworu kwasu chlorowodorowego. Ilość użytego kwasu powinna wynosić od 0,2 do 0,9 ml. Jednocześnie wykonuje się ślepą próbę, zastępując 50 ml próbki taką samą ilością wody.
5. OBLICZANIE WYNIKU
Zawartość lotnych zasad azotowych (Lz), w g/hl alkoholu etylowego 100% obj., wyrażoną w postaci azotu, oblicza się według wzoru:
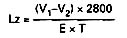
gdzie:
V1 – oznacza ilość ml kwasu chlorowodorowego użytego do zobojętnienia próbki podczas miareczkowania próbki,
V2 – oznacza ilość ml kwasu chlorowodorowego użytego w ślepej próbie,
T – oznacza zawartość alkoholu etylowego w próbce w procentach objętościowych,
E – oznacza ilość użytej próbki w mililitrach
6. POWTARZALNOŚĆ
Różnica między wynikami dwóch oznaczeń, przeprowadzonych jednocześnie lub w krótkim odstępie czasu, przez tego samego analityka, na tej samej próbce i w takich samych warunkach, nie może przekraczać 0,05 g/hl alkoholu etylowego 100% obj.
Załącznik nr 8
OZNACZANIE ZAWARTOŚCI SUCHEJ POZOSTAŁOŚCI PO ODPAROWANIU
Oznaczanie zawartości suchej pozostałości po jej odparowaniu polega na odparowaniu badanej próbki w łaźni wodnej i wysuszeniu pozostałości w suszarce w temperaturze 103 ± 20 C.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Próbka do analizy jest przechowywana w czystej kolbie zamkniętej doszlifowanym korkiem przemytym alkoholem lub korkiem zawiniętym w folię cynową lub aluminiową, w warunkach zapewniających brak dostępu powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
2. APARATURA
Do oznaczania zawartości suchej pozostałości po jej odparowaniu stosuje się:
1) łaźnię wodną umożliwiającą osiągnięcie temperatury wrzenia;
2) parownicę;
3) eksykator ze środkiem suszącym zawierającym świeżo aktywowany żel krzemionkowy (lub inny podobny środek suszący) ze wskaźnikiem wilgotności;
4) wagę analityczną;
5) suszarkę laboratoryjną o regulowanej temperaturze 103 ± 20 C.
3. WYKONANIE OZNACZANIA
Czystą suchą parownicę wazy się z dokładnością do 0,1 mg z próbki. Do parownicy odmierza się pipetą od 100 do 250 ml. Następnie parownicę z próbką umieszcza się we wrzącej łaźni wodnej i pozostawia ją do odparowania próbki.
Odparowaną próbkę umieszcza się na 30 minut w suszarce w temperaturze 103 ± 20 C, a następnie przenosi się do eksykatora i pozostawia do schłodzenia na 30 minut. Po ostudzeniu parownicę wraz z pozostałością wazy się z dokładnością do 0,1 mg.
4. OBLICZANIE WYNIKU
Zawartość suchej pozostałości po odparowaniu (Sm) w g/hl alkoholu etylowego 100% obj. oblicza się według wzoru:
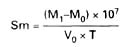
gdzie:
M0 – oznacza masę (g) suchej czystej parownicy,
M1 – oznacza masę (g) parownicy z pozostałością po suszeniu,
V0 – oznacza objętość (ml) próbki użytej do suszenia,
T – oznacza zawartość alkoholu etylowego w próbce w procentach objętościowych.
5. POWTARZALNOŚĆ
Różnica między wynikami dwóch oznaczeń, prze prowadzonych jednocześnie lub w krótkim odstępie czasu, przez tego samego analityka, na tej samej próbce i takich samych warunkach, nie może przekraczać 0,5 g/hl alkoholu etylowego 100% obj.
Załącznik nr 9
PRÓBA NA OBECNOŚĆ FURFURALU
Metoda polega na reakcji furfuralu z aniliną w środowisku kwaśnym. Powstanie łososiowo-różowego zabarwienia wskazuje na obecność furfuralu.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Próbka do analizy jest przechowywana w czystej kolbie zamkniętej doszlifowanym korkiem przemytym alkoholem lub korkiem zawiniętym w folię cynową lub aluminiową, w warunkach zapewniających brak dostępu powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
2. ODCZYNNIKI
Do wykonania próby na obecność furfuralu stosuje się następujące odczynniki chemiczne o czystości analitycznej:
1) anilinę świeżo destylowaną;
2) kwas octowy lodowaty.
3. APARATURA
Do wykonania próby na obecność furfuralu stosuje się probówki z doszlifowanymi szklanymi korkami.
4. WYKONANIE OZNACZANIA
Do probówki odmierza się pipetą 10 ml z próbki, następnie dodaje się 0,5 ml aniliny i 2 ml kwasu octowego lodowatego. Probówkę wstrząsa się w celu wymieszania jej zawartości.
5. INTERPRETACJA WYNIKU
Jeżeli czas pojawienia się zabarwienia łososiowo-różowego w probówce jest krótszy niż 20 minut, test ma wynik pozytywny i próbka zawiera furfural.
6. POWTARZALNOŚĆ
Wyniki dwóch testów na obecność furfuralu, przeprowadzonych jednocześnie lub w krótkim odstępie czasu przez tego samego analityka, na tej same; próbce i w takich samych warunkach powinny być identyczne.
Załącznik nr 10
TEST UV
Test UV polega na pomiarze absorbancji próbki w zakresie długości fal od 220 do 270 nm w stosunku do określonej substancji odniesienia o wysokiej czystości optycznej.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Próbka do analizy jest przechowywana w czystej kolbie zamkniętej doszlifowanym korkiem przemytym alkoholem lub korkiem zawiniętym w folię cynową lub aluminiową, w warunkach zapewniających brak dostępu powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
2. ODCZYNNIKI
Do wykonania testu UV stosuje się spektralnie czysty N-heksan.
3. APARATURA
Do wykonania testu UV stosuje się:
1) spektrofotometr UV-VIS, umożliwiający pomiar absorbancji przy długości fali od 220 do 270 nm;
2) kuwety kwarcowe o długości drogi optycznej 10 mm i tej samej transmisji widma.
4. WYKONANIE OZNACZANIA
Jedną kuwetę przepłukuje się n-heksanem, a następnie napełnia n-heksanem, drugą kuwetę przepłukuje się próbką i napełnia badaną próbką. Ścianki kuwet oczyszcza się z zewnątrz i wstawia do spektrofotometru. Mierzy się wartość absorbancji badanej próbki w porównaniu do n-heksanu przy długości fal od 220, 230, 240, 270 nm. Następnie wykreśla się krzywą w układzie A = f(λ), gdzie A oznacza wartość absorbancji, λ – długość fali.
5. OBLICZANIE WYNIKU
Wielkości absorbancji otrzymane dla 220, 230, 240, 270 nm nie mogą przekroczyć następujących wartości: 0,02; 0,08; 0,18 i 0,3.
Krzywa absorbancji powinna mieć przebieg płaski i regularny.
Załącznik nr 11
OZNACZANIE ZAWARTOŚCI IZOTOPU WĘGLA 14C
Oznaczanie zawartości izotopu węgla 14C umożliwia odróżnianie alkoholu syntetycznego od alkoholu etylowego rolniczego.
Naturalna zawartość izotopu węgla 14C w atmosferze (wartość odniesienia), asymilowana przez roślinność jest wartością zmienną. Wartość tę oznacza się w stosunku do alkoholu etylowego rolniczego z surowca pochodzącego z ostatniego sezonu wegetacyjnego.
Zawartość izotopu węgla 14C próbek alkoholowych jest wyznaczana bezpośrednio za pomocą cieczowego licznika scyntylacyjnego.
1. PRZYGOTOWANIE PRÓBKI DO ANALIZY
Próbka do analizy jest przechowywana w czystej kolbie zamkniętej doszlifowanym korkiem przemytym alkoholem lub korkiem zawiniętym w folię cynową lub aluminiową, w warunkach zapewniających brak dostępu powietrza i wilgoci. Należy przestrzegać, aby zamknięcie pojemnika nie miało kontaktu z próbką, i nie stosować żadnych substancji uszczelniających to zamknięcie.
Przed przystąpieniem do wykonania analizy pobraną próbkę miesza się w celu jej ujednolicenia.
Zawartość alkoholu etylowego rolniczego w badanej próbce powinna wynosić co najmniej 85% obj., próbka jest wolna od zanieczyszczeń, które absorbują przy długościach fal mniejszych niż 450 nm. Dopuszcza się niewielkie pozostałości estrów i aldehydów.
2. ODCZYNNIKI
Do oznaczania zawartości izotopu węgla 14C stosuje się:
1) toluen scyntylacyjny zawierający 5 g 2,5-dwufenyloaksozol (PPO) i 0,5 g p-bis-4-nrietylo-5-fenyloaksozoliol(2)-benzen (dwumetylo-POPOP) w 1 litrze toluenu o czystości analitycznej, przy czym dopuszcza się użycie gotowych, handlowych odczynników, o tym samym składzie;
2) wzorzec izotopu węgla 14C; 14C n-heksadekan o aktywności około 1 x 106 dpm/g (około 1,67 x 104 cBq/g) przy gwarantowanej dokładności oznaczania tej aktywności wynoszącej ± 2%;
3) alkohol etylowy rolniczy bez izotopu węgla 14C, tj. alkohol syntetyczny z surowców kopalnych zawierający co najmniej 85% masy alkoholu etylowego, do oznaczania tła;
4) alkohol etylowy rolniczy zawierający co najmniej 85% masy alkoholu etylowego, stanowiący materiał odniesienia.
3. APARATURA
Do oznaczania zawartości izotopu węgla 14C stosuje się:
1) wielokanałowy scyntylacyjny spektrofotometr cieczowy z procesorem i automatycznym wzorcowaniem zewnętrznym oraz odczytem zewnętrznego wskaźnika wzorzec/kanał (standardowa konstrukcja: trzy kanały metrowe oraz dwa wzorcowe kanały zewnętrzne);
2) niskopotasowe kuwety nadające się do spektrofotometru, z ciemnymi nakrętkami śrubowymi zawierającymi wkładki polietylenowe;
3) pipety pomiarowe o pojemności 10 ml;
4) automatyczny dozownik o pojemności 10 ml;
5) kolbę kulistą o pojemności 250 ml z korkiem z matowego szkła;
6) aparat do destylacji alkoholowej z płaszczem grzejącym;
7) strzykawkę mikrolitrową o pojemności 50 ul;
8) lejek do piknometrów, piknometry o pojemności 25 ml i 50 ml;
9) termostat ze stabilizacją temperatury ± 0,010 C;
10) Tablice Alkoholometryczne.
4. WYKONANIE OZNACZANIA
Uruchomienie sprzętu
Sprzęt ustawia się zgodnie z instrukcją producenta. Optymalne warunki pomiaru uzyskuje się wtedy, gdy wartość E/B – wskaźnik jakości, ma wartość maksymalną,
gdzie:
E – oznacza skuteczność,
B – oznacza tło.
Optymalizowane są jedynie dwa kanały metrowe. Trzeci jest pozostawiony otwarty dla celów kontrolnych.
Wybór kuwet
Kuwety napełnia się 10 ml syntetycznego alkoholu etylowego rolniczego bez zawartości 14C oraz 10 ml toluenu scyntylacyjnego. Dokonuje się pomiaru dla każdej z kuwet. Czas pomiaru wynosi co najmniej 2 x 100 minut. Odrzuca się te kuwety, których tła różnią się od średniej o więcej niż ± 1%. Używa się wyłącznie kuwet nowych pochodzących z tej samej partii towaru.
Oznaczanie zewnętrznego stosunku wzorzec/kanał (ESCR)
Podczas procesu ustawiania kanałów, po oznaczeniu skuteczności, oznacza się ESCR za pomocą programu komputerowego. Używany wzorzec zewnętrzny to 137cez, który jest fabrycznie wbudowany przez producenta.
Przygotowanie próbki
Dopuszcza się wykonywanie pomiaru próbek o zawartości co najmniej 85% masowych, wolnych od zanieczyszczeń, które absorbują przy długościach fal mniejszych niż 450 nm. Po odrzuceniu pierwszych kilku ml próbka jest destylowana bezpośrednio do piknometru, po czym metodą piknometryczną oznacza się zawartość alkoholu w próbce. Zawartości alkoholu odczytuje się z Tablic Alkoholometrycznych.
Wykonanie pomiaru za pomocą wzorca zewnętrznego
Odmierza się pipetą 10 ml z każdej próbki do wybranej kuwety o sprawdzonym tle, po czym za pomocą automatycznego dozownika dodaje się 10 ml toluenu scyntylacyjnego. Próbki w kuwetach są mieszane ruchami obrotowymi w taki sposób, by nie dopuścić do nawilżania przez ciecz wkładki polietylenowej w tle.
Aby sprawdzić roczną wartość izotopu węgla 14C przygotowuje się duplikat alkoholu etylowego fermentacyjnego otrzymanego z surowców pochodzących z ostatniego sezonu wegetacyjnego, następnie zawartość kuwety miesza się z wewnętrznym wzorcem.
Próbki kontrolne i próbki tła umieszcza się na początku szeregu pomiarowego. Każdy z nich powinien zawierać nie więcej niż 10 próbek do analizy. Całkowity czas pomiaru jednej próbki wynosi co najmniej 2 x 100 minut, przy czym poszczególne próbki mierzy się etapami po 100 minut w celu wyeliminowania błędu (jeden cykl odpowiada przedziałowi czasu 100 minut na próbkę).
Próbki tła i próbki kontrolne powinny być przygotowywane co cztery tygodnie.
W przypadku próbek o słabej absorpcji (ESCR około 1,8) wartością ma jedynie nieznaczny wpływ na skuteczność analizy. Jeżeli zmiana mieści się w granicach + 5%, można spodziewać się podobnej skuteczności. W przypadku próbek o silniejszej absorpcji, takich jak alkohole etylowe rolnicze denaturowane, skuteczność można ustalić za pomocą korekcyjnego wykresu wygaszania. W przypadku braku programu komputerowego używa się wzorca wewnętrznego w celu uzyskania wyników.
Pomiar próbek przy pomocy wewnętrznego wzorca heksadekanu 14C
Próbki kontrolne i próbki tła (alkohol fermentacyjny i syntetyczny) oraz materiał nieznany są poddawane pomiarom jako duplikaty. Jedna próbka duplikatu przygotowywana jest w nieselektywnej kuwecie, a jako wzorzec wewnętrzny dodaje się dokładnie odmierzoną ilość (30 ul) heksadekanu 14C o dodatkowej aktywności około 26 269 dpm/g C (około 43 782 cBq/g C). Przygotowania próbek i czasu pomiaru pozostałych próbek dokonuje się w sposób, o którym mowa w pkt 2. Czas pomiaru próbek ze wzorcem wewnętrznym można ograniczyć do około pięciu minut poprzez wstępne ustawienie na 105 impulsów. Do serii pomiarowej używa się jednego duplikatu z każdej próbki kontrolnej i próbki tła. Są one umieszczane na początku szeregu pomiarowego. Aby zapobiec zanieczyszczeniom podczas pomiaru z wzorcem wewnętrznym, próbki należy przetrzymywać z dala od miejsca przygotowywania i pomiaru próbek do analizy. Po dokonaniu pomiaru kuwety sprawdzone pod względem tła mogą być użyte ponownie. Wyrzuca się nakrętki i kuwety zawierające wzorzec wewnętrzny.
5. OBLICZANIE WYNIKU
Jednostką aktywności substancji radioaktywnej jest bekerel (Bq).
Wskaźnik radioaktywności właściwej wyrażony w bekerelach w stosunku do jednego grama węgla (Bq/g C).
Dopuszcza się wyrażanie wyniku w centybekerelach (cBq/g C).
Wskaźnik radioaktywności właściwej oblicza się według wzorów:
1) w przypadku zastosowania wzorca zewnętrznego:
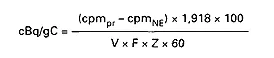
2) w przypadku zastosowania wzorca wewnętrznego:
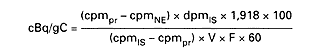
gdzie:
| cpmPR | - oznacza średni wskaźnik licznika podczas całego czasu pomiaru próbki, |
| cpmNE | - oznacza średni wskaźnik impulsów tła obliczony w ten sam sposób, |
| cpmis | - oznacza ilość dodatkową dodanego wzorca wewnętrznego (radioaktywność wzorcowania dpm), |
| dpmis | - oznacza ilość dodanego wzorca wewnętrznego (radioaktywność wzorcowania dpm), |
| V | - oznacza objętość w ml użytych próbek, |
| F | - oznacza zawartość w gramach czystego alkoholu na ml odpowiadającą jego stężeniu, |
| Z | - oznacza skuteczność odpowiadającą wartości ESCR 918 = ilość gramów alkoholu na gram węgla. |
6. POWTARZALNOŚĆ
Powtarzalność jest to wartość, od której jest mniejsza wartość bezwzględna różnicy między dwoma pojedynczymi wynikami badań, uzyskanymi z zastosowaniem tej samej metody, dla tej samej próbki, w tym samym laboratorium, przez tego samego analityka, z użyciem tego samego sprzętu, w krótkim odstępie czasu.
W przypadku oznaczania zawartości izotopu węgla 14C powtarzalność wynosi:
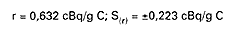
7. ODTWARZALNOŚĆ
Odtwarzalność jest to wartość, od której jest mniejsza wartość bezwzględna różnicy między dwoma pojedynczymi wynikami badań, uzyskanymi z zastosowaniem tej samej metody, dla tej samej próbki, w różnych laboratoriach, przez różnych analityków, z użyciem różnego sprzętu.
W przypadku oznaczania zawartości izotopu węgla 14C odtwarzalność wynosi:
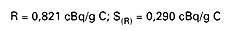
- Data ogłoszenia: 2003-08-07
- Data wejścia w życie: 2003-08-22
- Data obowiązywania: 2003-08-22
- Dokument traci ważność: 2006-12-22

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA