REKLAMA
Dziennik Ustaw - rok 2003 nr 105 poz. 996
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 30 maja 2003 r.
w sprawie sposobu przedstawiania dokumentacji oraz wzoru wniosku o dopuszczenie do obrotu produktu homeopatycznego i produktu homeopatycznego weterynaryjnego
Na podstawie art. 21 ust. 8 i 9 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne (Dz. U. Nr 126, poz. 1381, z późn. zm.2)) zarządza się, co następuje:
Minister Zdrowia: L. Sikorski
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 28 czerwca 2002 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 93, poz. 833).
2) Zmiany wymienionej ustawy zostały ogłoszone w Dz. U. z 2002 r. Nr 113, poz. 984, Nr 141, poz. 1181 i Nr 152, poz. 1265 oraz z 2003 r. Nr 45, poz. 391.
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 30 maja 2003 r. (poz. 996)
Załącznik nr 1
SZCZEGÓŁOWY SPOSÓB PRZEDSTAWIANIA DOKUMENTACJI
1. Wniosek o dopuszczenie do obrotu produktu homeopatycznego, w tym produktu homeopatycznego weterynaryjnego, zwanego dalej „produktem”, powinien zawierać informacje zgodne z aktualnym stanem wiedzy oraz powinien być wypełniony czytelnie.
2. W dokumentacji stosuje się nazwy naukowe lub farmakopealne zgodnie z obowiązującą terminologią.
W przypadku:
1) substancji homeopatycznych czynnych, które są zamieszczone w Farmakopei Polskiej, Farmakopei Europejskiej, farmakopei homeopatycznej lub w farmakopei uznawanej w państwach członkowskich Unii Europejskiej – główny tytuł monografii, która będzie obligatoryjna dla wszystkich takich substancji, ze wskazaniem farmakopei;
2) substancji innych niż homeopatyczne – nazwy międzynarodowe (INN) w wersji angielskiej, zalecane przez Światową Organizację Zdrowia (WHO), którym mogą towarzyszyć inne niezastrzeżone nazwy lub jeżeli ich nie ma, dokładny opis naukowy; substancje nieposiadające międzynarodowej nazwy ani dokładnego opisu naukowego należy określić, opisując, w jaki sposób i z czego zostały przygotowane, o jakie składniki wzbogacone, oraz podać inne ważne informacje;
3) barwników – oznaczenie kodem „E” określonym w przepisach wydanych na podstawie art. 27 ust. 2 ustawy i dnia 6 września 2001 r. – Prawo farmaceutyczne.
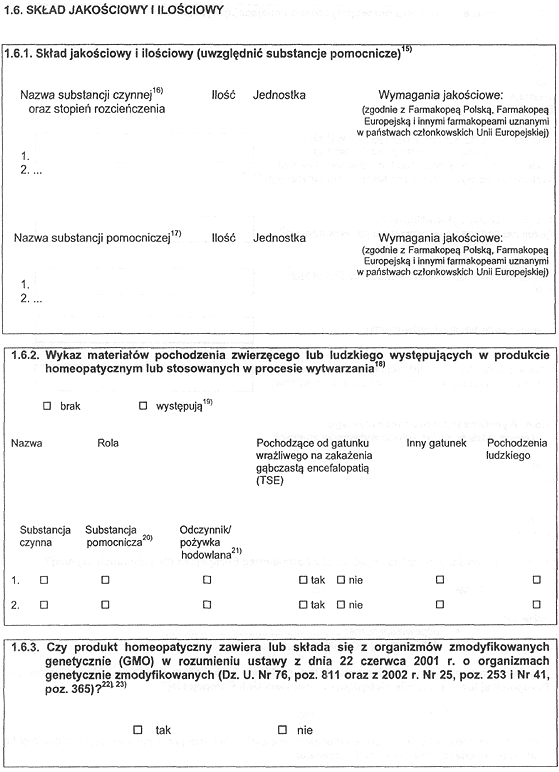
3. Dokumentacja powinna zawierać informację o składzie jakościowym i ilościowym produktu, a w szczególności o:
1) substancji czynnej; w celu podania opisu ilościowego substancji czynnych zawartych w produkcie homeopatycznym należy uwzględnić symbol rozcieńczenia: D lub DH, C lub CH, X lub XH, K, LM (Q), TM; ilość substancji czynnych należy wyrazić w jednostkach masy przypadającej na jednostkę dawkowania lub na jednostkę masy bądź objętości produktu;
2) rozpuszczalnikach, niezależnie od ich rodzaju lub użytych ilości, oraz substancjach konserwujących, stabilizujących, emulgujących, aromatycznych, barwnikach i markerach;
3) substancjach pomocniczych wchodzących w skład postaci farmaceutycznej, niezależnie od ich rodzaju i ilości; ilość substancji pomocniczych należy wyrazić w jednostkach masy przypadających na jednostkę dawkowania lub na jednostkę masy bądź objętości produktu.
4. Dokumentacja powinna zawierać dane dotyczące warunków przechowywania i transportu zgodnie z wynikami badań dotyczących trwałości produktu, które powinny być opracowane zgodnie z wymogami obowiązującymi w państwach członkowskich Unii Europejskiej.
5. Dokumentacja dotycząca wielkości i rodzaju opakowania zawiera także dane dotyczące opakowania i, jeżeli to celowe, sposobu jego zamknięcia oraz informacje dotyczące urządzeń, za pomocą których produkt będzie podawany, a które będą do niego dołączone.
6. Dokumenty dołączone do wniosku powinny zawierać opis sposobu otrzymywania i kontroli roztworu macierzystego:
1) homeopatycznej substancji czynnej nieopisanej w Farmakopei Polskiej, Farmakopei Europejskiej, farmakopei homeopatycznej lub w farmakopei uznawanej w państwach członkowskich Unii Europejskiej;
2) substancji czynnej opisanej w Farmakopei Polskiej, Farmakopei Europejskiej, farmakopei homeopatycznej lub w farmakopei uznawanej w państwach członkowskich Unii Europejskiej, jeżeli została przygotowana metodą mogącą pozostawić zanieczyszczenia, które nie zostały zamieszczone w monografii farmakopealnej i dla której monografia nie jest właściwa, aby odpowiednio kontrolować jej jakość.
7. Szczegółowy sposób otrzymania i kontroli jakości roztworu macierzystego w czasie produkcji i procesu walidacji może być przekazany przez wytwórcę substancji czynnych do Prezesa Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych, zwanego dalej „Prezesem Urzędu”. Wytwórca powinien przekazać podmiotowi odpowiedzialnemu pisemne potwierdzenie, że zapewni zgodność wszystkich serii produktu oraz nie dokona zmian procesu produkcji lub dokumentacji bez powiadamiania podmiotu odpowiedzialnego. W przypadku wprowadzenia jakichkolwiek zmian należy przekazać Prezesowi Urzędu dokumentację uzasadniającą te zmiany.
8. Dokumenty dołączone do wniosku powinny zawierać wyniki kontroli jakości wszystkich użytych materiałów wyjściowych, przy czym:
1) do dokumentów materiałów wyjściowych opisanych w farmakopeach dołącza się:
a) monografie farmakopei homeopatycznej, Farmakopei Europejskiej i Farmakopei Polskiej do wszystkich substancji w niej opisanych,
b) rutynowe testy przeprowadzone na każdej serii materiałów wyjściowych zgodne z danymi zawartymi we wniosku o dopuszczenie do obrotu; w przypadku użycia testów innych niż opisane w farmakopei należy złożyć oświadczenie, że materiały wyjściowe spełniają wymagania jakościowe zawarte w farmakopei,
c) dokument potwierdzający odpowiednią jakość materiału wyjściowego, jeżeli dokumentacja lub inne postanowienia określone w monografii Farmakopei Polskiej, Farmakopei Europejskiej, farmakopei homeopatycznej lub farmakopei uznawanej przez państwa członkowskie Unii Europejskiej są niewystarczające do zapewnienia jakości substancji,
d) kopię monografii farmakopei, włącznie z walidacją procedur wykonania testu zawartą w farmakopei oraz tłumaczeniem monografii farmakopei na język polski lub angielski; jeżeli materiał wyjściowy nie jest opisany w Farmakopei Polskiej, Farmakopei Europejskiej ani w farmakopei uznawanej przez państwa członkowskie Unii Europejskiej, można odwołać się do innej farmakopei;
2) do dokumentów materiałów wyjściowych nieopisanych w farmakopeach dołącza się:
a) dla biologicznych materiałów wyjściowych:
– opis przedstawiony w formie monografii,
– informacje dotyczące wszystkich substancji biologicznych użytych w procesie produkcji,
– informacje dotyczące źródła pochodzenia materiałów,
– informacje dotyczące każdej obróbki, zastosowanych metod oczyszczania i inaktywacji, łącznie z informacją dotyczącą walidacji tych procesów i kontroli etapów pośrednich,
– informacje dotyczące wszystkich przeprowadzonych testów wykrywających zanieczyszczenia uboczne, które są wykonywane dla każdej serii substancji, jeżeli dotyczy,
b) dla niebiologicznych materiałów wyjściowych:
– opis w formie monografii, z zastosowaniem nazwy materiału wyjściowego, uzupełnionej o synonimy handlowe lub naukowe,
– opis wytwarzania materiału wyjściowego zgodny z monografią ogólną zawartą w Farmakopei Polskiej, Farmakopei Europejskiej, farmakopei homeopatycznej,
– metody identyfikacji i oznaczania zawartości substancji czynnych, jeżeli dotyczy,
– opis czystości, z uwzględnieniem ogólnej ilości przewidywanych zanieczyszczeń, szczególnie tych, które mogą wywierać efekt szkodliwy, a jeżeli to konieczne – także tych, które odnoszą się do kombinacji substancji, których dotyczy wniosek, a które mogą mieć działanie uboczne na trwałość produktu lub mogą zmieniać wyniki analiz; dołącza się opis testów, które są przeprowadzane w celu ustalenia czystości każdej serii materiału wyjściowego, jeżeli dotyczy,
– informacje o szczególnych środkach ostrożności wymaganych w czasie przechowywania materiału wyjściowego, a jeżeli to konieczne – o czasie jego przechowywania.
9. Opis procesu wytwarzania, w tym opis sposobu rozcieńczania i dynamizacji, powinien zawierać:
1) etapy produkcji, w tym etapy rozcieńczania i dynamizacji, zgodnie z farmakopeą homeopatyczną, opisane w taki sposób, aby można było dokonać oceny powtarzalności procesu produkcyjnego i ryzyka wystąpienia w produkcie końcowym efektów ubocznych, w tym zanieczyszczenia mikrobiologicznego;
2) wykaz substancji, które ulegają usunięciu w czasie procesu wytwarzania;
3) określenie etapu produkcji, w którym pobiera się próbki w celu przeprowadzenia kontroli etapów pośrednich;
4) dane dotyczące walidacji procesu wytwarzania;
5) wskazanie przepisu farmakopei homeopatycznej, według którego wytwarzany jest macierzysty roztwór homeopatyczny oraz produkt homeopatyczny;
6) dane dotyczące badań kontrolnych, które są przeprowadzane na etapach pośrednich.
10. Dokumenty dołączone do wniosku powinny zawierać opis metod kontroli formy farmaceutycznej, w tym badania stabilności i czystości mikrobiologicznej, przy czym:
1) opis badań kontrolnych formy farmaceutycznej powinien zawierać:
a) jeżeli istnieją odpowiednie monografie, a zastosowane są procedury i zakresy wykonania testów inne niż zawarte w monografiach Farmakopei Polskiej, Farmakopei Europejskiej, farmakopei homeopatycznej, należy dołączyć potwierdzenie, że produkt końcowy badany zgodnie z opisem zawartym w monografii będzie spełniał wymagania jakościowe dotyczące postaci farmaceutycznej opisanej w monografii. Wniosek o dopuszczenie do obrotu zawiera wyszczególnienie badań kontrolnych formy farmaceutycznej, które są przeprowadzane na reprezentatywnych próbach każdej serii produktu gotowego,
b) specyfikacje formy farmaceutycznej oraz opis metod kontroli,
c) badanie jakościowe i ilościowe substancji czynnej, jeżeli dotyczy,
d) ustalony opis metod analizy produktu gotowego, w tym szczegółowe opisy umożliwiające łatwe odtworzenie metod,
e) specyficzne testy identyfikacyjne, jeżeli dotyczy,
f) badanie tożsamości rozpuszczalników, jeżeli dotyczy,
g) metodę wykonania testu zaproponowanego do identyfikacji barwników,
h) oznaczenie ilościowe konserwantów, jeżeli dotyczy,
i) badania jałowości i czystości mikrobiologicznej.
W przypadkach gdy oznaczenie homeopatycznych substancji czynnych obecnych w małych ilościach wymagałoby skomplikowanych oznaczeń, trudnych do przeprowadzenia w odniesieniu do każdej serii produkcyjnej, należy przyjąć jako wystarczający pełen opis procesu wytwarzania;
2) do potwierdzenia okresu ważności przechowywania roztworu macierzystego i produktu końcowego przedstawia się opis zastosowanych badań stabilności farmaceutycznej, biologicznej i chemicznej. Badania powinny być wykonane w czasie rzeczywistym. Należy je przeprowadzić na wystarczającej liczbie serii produktu w postaci gotowej do sprzedaży;
3) w celu udokumentowania, że jakość produktu homeopatycznego jest odpowiednia, należy dostarczyć walidację metod analitycznych. W przypadku ograniczenia badań należy to udokumentować, jeżeli dotyczy;
4) podmiot odpowiedzialny wykazuje, że produkt homeopatyczny jest wytwarzany metodami mającymi na celu zmniejszenie ryzyka przenoszenia czynników zakaźnych gąbczastych encefalopatii zwierzęcych, uwzględniając w tym zakresie stan wiedzy naukowej w chwili składania wniosku.
11. Do wniosku należy dołączyć projekty etykiet i ulotki informacyjnej, jeżeli dotyczy.
12. Dokumenty dołączone do wniosku powinny zawierać dane dotyczące opakowania bezpośredniego, z podaniem wymagań jakościowych, oraz wzory opakowań bezpośrednich i zewnętrznych, jeżeli dotyczy.
13. Dokumenty dołączone do wniosku powinny zawierać wszystkie dane dotyczące bezpieczeństwa produktu homeopatycznego.
W przypadku gdy surowiec homeopatyczny lub roztwór macierzysty jest znany i opisany w literaturze homeopatycznej, a w szczególności w: Materia Medica – Boericke, Charette, Clarke, Kent, Leser, Mezger, Staufer, Voision, można powołać się na literaturę fachową.
14. W przypadku produktów stosowanych u zwierząt, których tkanki lub produkty przeznaczone są do spożycia, dokumentacja powinna zawierać wszystkie dane dotyczące bezpieczeństwa produktu homeopatycznego weterynaryjnego w celu uniknięcia ryzyka dla zdrowia konsumenta żywności pochodzącej od leczonych zwierząt lub uniknięcia zaburzeń w procesach technologicznych.
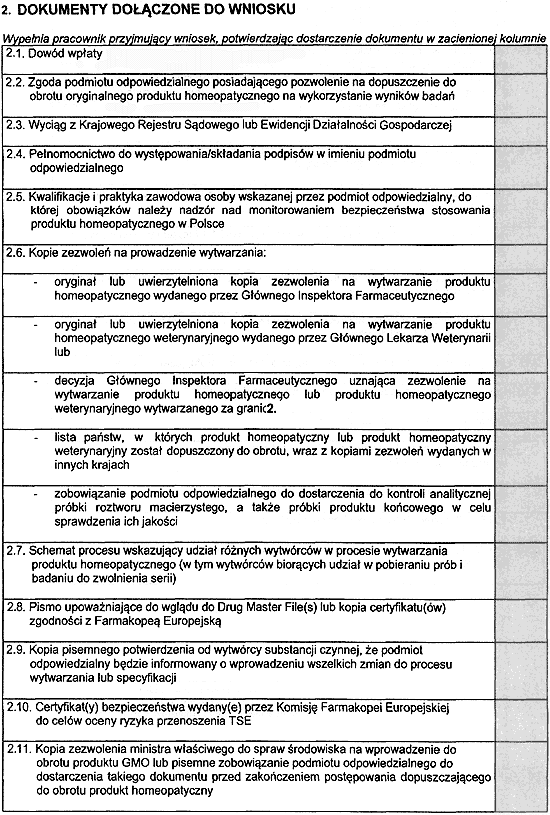
15. Do wniosku należy dołączyć:
1) oryginał lub uwierzytelnioną kopię zezwolenia na wytwarzanie produktu homeopatycznego wydanego przez Głównego Inspektora Farmaceutycznego;
2) oryginał lub uwierzytelnioną kopię zezwolenia na wytwarzanie produktu homeopatycznego weterynaryjnego wydanego przez Głównego Lekarza Weterynarii lub
3) decyzję Głównego Inspektora Farmaceutycznego uznającą zezwolenie na wytwarzanie produktu homeopatycznego lub produktu homeopatycznego weterynaryjnego wytwarzanego za granicą;
4) listę państw, w których produkt homeopatyczny lub produkt homeopatyczny weterynaryjny został dopuszczony do obrotu, wraz z kopiami zezwoleń wydanych w innych krajach;
5) zobowiązanie podmiotu odpowiedzialnego do dostarczenia do kontroli analitycznej próbki roztworu macierzystego, a także próbki produktu końcowego w celu sprawdzenia ich jakości.
Załącznik nr 2












- Data ogłoszenia: 2003-06-17
- Data wejścia w życie: 2003-07-02
- Data obowiązywania: 2003-07-02

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA