REKLAMA
Dziennik Ustaw - rok 2002 nr 241 poz. 2098
ROZPORZĄDZENIE MINISTRA ZDROWIA1)
z dnia 24 grudnia 2002 r.
w sprawie warunków bezpiecznego stosowania promieniowania jonizującego w celach medycznych oraz sposobu wykonywania kontroli wewnętrznej nad przestrzeganiem tych warunków.
Na podstawie art. 15 ust. 3 ustawy z dnia 29 listopada 2000 r. – Prawo atomowe (Dz. U. z 2001 r. Nr 3, poz. 18, Nr 100, poz. 1085 i Nr 154, poz. 1800 oraz z 2002 r. Nr 74, poz. 676 i Nr 135, poz. 1145) zarządza się, co następuje:
Rozdział 1
Przepisy ogólne
§ 1.
1) „audyt kliniczny” – systematyczną kontrolę lub przegląd medycznych procedur radiologicznych, mające na celu polepszenie jakości otrzymywanych przez pacjenta świadczeń zdrowotnych poprzez usystematyzowaną analizę, w ramach której praktyka, procedury i wyniki radiologiczne są porównywane z uznanymi standardami oraz, w razie konieczności, modyfikację dotychczasowego postępowania lub wprowadzenie nowych standardów;
2) „dawka tolerancji” – dopuszczalną dawkę promieniowania jonizującego pochłoniętą przez określony narząd lub tkankę zdrową lub ich część, której odpowiada akceptowane ryzyko późnego powikłania popromiennego wynoszące nie więcej niż 5% w okresie 5 lat po leczeniu, z wyjątkiem rdzenia kręgowego i ośrodkowego układu nerwowego, dla których nie może przekroczyć 1%;
3) „fizyk medyczny” – specjalistę w dziedzinie fizyki lub technologii promieniowania w odniesieniu do ekspozycji na promieniowanie jonizujące w celach medycznych;
4) „istotny błąd dozymetryczny” – niezamierzoną różnicę (5–25%) między całkowitą przepisaną dawką promieniowania a dawką rzeczywiście zaaplikowaną w trakcie leczenia radioterapeutycznego, zwiększającą prawdopodobieństwo nieakceptowanego wyniku leczenia, poprzez wystąpienie powikłań, ale niezwiększającą w znacznym stopniu ryzyka utraty życia przez pacjenta poddawanego radioterapii;
5) „kontrola jakości” – zespół działań wchodzących w skład zarządzania jakością, polegających na kontroli planowania, koordynacji i realizacji, mających na celu utrzymanie lub poprawę jakości funkcjonowania urządzeń radiologicznych oraz procedur diagnostycznych i leczniczych; do kontroli jakości należy w szczególności ocena i utrzymanie wymaganych wartości dla wszystkich parametrów eksploatacyjnych urządzeń radiologicznych;
6) „medyczna procedura radiologiczna” – procedurę medyczną polegającą na ekspozycjach, o których mowa w art. 15 ust. 1 ustawy z dnia 29 listopada 2000 r. – Prawo atomowe, zwanej dalej „ustawą”
7) „medycyna nuklearna” – wszelką działalność diagnostyczną związaną z podawaniem pacjentom produktów radiofarmaceutycznych, a także z zabiegami terapeutycznymi przy użyciu produktów radiofarmaceutycznych;
8) „narządy krytyczne” – narządy wewnętrzne człowieka, których obecność w objętości tarczowej lub w jej pobliżu ma wpływ na wartość planowanej dawki w radioterapii;
9) „objętość tarczowa” – objętość guza nowotworowego lub innych tkanek, które są napromieniane w celu osiągnięcia planowanego efektu terapeutycznego;
10) „odpowiedzialność kliniczna” – odpowiedzialność, jaka spoczywa na lekarzu realizującym procedury prowadzące do indywidualnej ekspozycji na promieniowanie jonizujące w celach medycznych; obejmuje ona w szczególności: uzasadnienie, optymalizację ochrony przed promieniowaniem, kliniczną ocenę wyniku, współpracę z innymi specjalistami i personelem, a w razie potrzeby – uzyskiwanie informacji o wynikach poprzednich badań, a także przekazywanie informacji lub dokumentacji radiologicznej innym lekarzom i ewentualne informowanie pacjenta oraz innych zainteresowanych osób realizujących medyczne procedury radiologiczne o ryzyku związanym ze stosowaniem promieniowania jonizującego;
11) „osoba kierująca na badanie lub zabieg” – lekarza, lekarza stomatologa lub felczera, uprawnionych, na podstawie odrębnych przepisów, do kierowania pacjenta na zabieg związany z ekspozycją na promieniowanie w celach medycznych;
12) „osoba wykonująca badanie lub zabieg” – lekarza, lekarza stomatologa lub inną osobę należącą do personelu medycznego, która ma uprawnienia do ponoszenia odpowiedzialności za ekspozycję pacjenta na promieniowanie w celach medycznych;
13) „poziomy referencyjne” – dawki promieniowania jonizującego na powierzchnię skóry w badaniach rentgenodiagnostycznych lub poziomy aktywności – w przypadku podawania pacjentom produktów radiofarmaceutycznych, dotyczące badań typowych pacjentów, dla poszczególnych kategorii urządzeń radiologicznych; poziomy referencyjne nie mogą być przekraczane w przypadku powszechnie stosowanych medycznych procedur radiologicznych, jeżeli stosuje się właściwe sposoby postępowania i urządzenia techniczne; poziomy referencyjne mogą być przekraczane w przypadku istnienia istotnych wskazań klinicznych;
14) „radioterapia” – wszelką działalność terapeutyczną związaną z wykorzystaniem aparatów terapeutycznych emitujących fotonowe promieniowanie jonizujące lub cząstki, urządzenia do terapii hadronowej, a także źródła izotopowe wprowadzane bezpośrednio do narządów wewnętrznych śródtkankowo, dojamowo, a także umieszczane na powierzchni ciała pacjenta (brachyterapia);
15) „rentgenodiagnostyka” – wszelką działalność diagnostyczną związaną z wykorzystaniem konwencjonalnych aparatów rentgenowskich, mammografów, tomografów komputerowych, rentgenowskich densytometrów kości, a także zabiegi wykonywane pod kontrolą aparatu rentgenowskiego, zwane dalej „zabiegami z zakresu radiologii interwencyjnej”
16) „ryzyko radiacyjne” – wielkość zagrożenia lub prawdopodobieństwo szkodliwego, a w szczególności chorobowego, następstwa związanego z dokonaną lub potencjalną ekspozycją na promieniowanie jonizujące; obejmuje ono zarówno prawdopodobieństwo pojawienia się niepożądanych następstw, jak i ich nasilenie oraz charakter;
17) „urządzenie radiologiczne” – urządzenie zawierające źródło promieniowania jonizującego lub służące do detekcji promieniowania, w tym w szczególności kamery scyntylacyjne i pozytronowy tomograf emisyjny PET, wykorzystywane do celów leczniczych lub diagnostycznych;
18) „wypadek w radioterapii” – efekt kliniczny i patologiczny wynikający z niezamierzonej różnicy, większej niż 25%, między całkowitą przepisaną dawką promieniowania a dawką rzeczywiście zaaplikowaną w trakcie leczenia radioterapeutycznego, zwiększający w znacznym stopniu ryzyko ciężkich powikłań u pacjenta z utratą życia włącznie lub spadku wyleczalności nowotworu, gdy dawka zaaplikowana jest mniejsza niż przepisana o więcej niż 25%; wypadkiem w radioterapii jest również napromienienie niewłaściwego pacjenta, błędna anatomicznie lokalizacja obszaru napromienienia oraz niewłaściwy rozkład dawki, w tym przy użyciu nieprawidłowego typu wiązki lub energii fotonu lub cząstek, a także niewłaściwe frakcjonowanie, jeżeli prowadzą one do nieosiągnięcia założonych efektów terapeutycznych lub odległych w czasie ciężkich następstw zdrowotnych;
19) „zarządzanie jakością” – zespół systematycznie planowanych i wykonywanych działań, koniecznych dla wystarczającego zapewnienia, że dana struktura, system lub ich części składowe bądź procedury będą działać w sposób zadowalający, spełniając właściwe normy.
2. Dopuszczenie do powszechnego stosowania medycznych procedur radiologicznych wymaga spełnienia warunków, o których mowa w ust. 1.
3. Medyczne procedury radiologiczne podlegają opiniowaniu (ocenie) właściwego konsultanta krajowego w przypadku uzyskania nowych danych dotyczących ich skuteczności i możliwych następstw zdrowotnych.
4. Uzasadnienie ekspozycji pacjenta na promieniowanie jonizujące w celach medycznych następuje przed jej wykonaniem, z uwzględnieniem celów badania lub leczenia oraz cech charakterystycznych pacjenta, którego ekspozycja dotyczy.
5. Jeżeli medyczna procedura radiologiczna nie została uzasadniona w sposób, o którym mowa w ust. 1, wykonanie badania lub zabiegu indywidualnego może być uzasadnione w szczególnych sytuacjach, podlegających w każdym przypadku odrębnej ocenie osoby wykonującej badanie lub zabieg. Dotyczy to w szczególności ekspozycji w celach prawno-medycznych.
6. Mogą istnieć okoliczności wykluczające lub zmniejszające zakres stosowania medycznej procedury radiologicznej u pacjenta, pomimo że procedura ta jest uznawana za ogólnie uzasadnioną. O zakresie stosowania medycznej procedury radiologicznej decyduje lekarz.
7. Dokonywanie ekspozycji na promieniowanie jonizujące w celach medycznych, które nie zostały uprzednio uzasadnione w sposób, o którym mowa w ust. 1, 5 lub 6, jest niedopuszczalne.
2. Skierowanie, o którym mowa w ust. 1, może być wystawione po upewnieniu się, że inne alternatywne i nieinwazyjne metody lub wcześniej wykonane badania radiologiczne nie mogą dostarczyć niezbędnych, analogicznych informacji lub wyników leczniczych.
3. Skierowanie, o którym mowa w ust. 1, w formie pisemnej lub elektronicznej, zawiera:
1) cel i uzasadnienie badania lub zabiegu;
2) wstępne rozpoznanie kliniczne;
3) informacje niezbędne do prawidłowego przeprowadzenia medycznej procedury radiologicznej;
4) podpis osoby kierującej na badanie lub zabieg.
4. Bez skierowania może być wykonane badanie z zastosowaniem promieniowania jonizującego przeprowadzane w ramach badań przesiewowych lub stomatologicznych – z wyłączeniem badań pantomograficznych – oraz w przypadkach bezpośredniego zagrożenia życia pacjenta.
2. Odpowiedzialność kliniczną za badanie lub zabieg z zastosowaniem promieniowania jonizującego ponosi lekarz specjalista w dziedzinie radioterapii lub medycyny nuklearnej, który planował, nadzorował i prowadził takie leczenie.
3. Przerwanie lub zaniechanie podjętego badania lub zabiegu z zastosowaniem promieniowania jonizującego może podjąć lekarz, o którym mowa w ust. 2, po zasięgnięciu konsyliarnej opinii w przypadkach:
1) stwierdzonej błędnej kwalifikacji do leczenia;
2) błędu w zakresie fizycznych lub technicznych parametrów napromieniania stwierdzonego w trakcie leczenia;
3) nietolerancji leczenia lub zagrożenia życia pacjenta przez kontynuowanie terapii.
4. Lekarz przeprowadzający badanie lub zabieg z zastosowaniem promieniowania jonizującego, po jego zakończeniu, informuje o przebiegu tego badania lub zabiegu osobę kierującą na leczenie lub zabieg.
2. Wartości dawek promieniowania lub aktywności produktu radiofarmaceutycznego przy badaniach i zabiegach, o których mowa w ust. 1, nie mogą, z zastrzeżeniem ust. 4, przekraczać poziomów referencyjnych, o których mowa w ust. 3.
3. Poziomy referencyjne dla badań z zastosowaniem promieniowania jonizującego lub zabiegów z zakresu radiologii określają załączniki nr 1 i 2 do rozporządzenia.
4. Przekroczenie poziomów referencyjnych, o których mowa w ust. 3, może być uzasadnione wyłącznie istotnymi wskazaniami klinicznymi oraz szczególnymi warunkami wykonywania badań lub zabiegów, o których mowa w ust. 1.
5. Dawki dla osób poddawanych ekspozycji w celach prawno-medycznych powinny być możliwie jak najmniejsze.
6. Za właściwe wykonanie badań i zabiegów, o których mowa w ust. 1, oraz za ograniczenie do minimum ekspozycji pacjenta na promieniowanie jonizujące odpowiada lekarz, który je wykonuje lub nadzoruje.
1) posiadających specjalizację w zakresie dyscyplin, w których są one stosowane;
2) którzy odbyli staż w zakresie technik interwencyjnych, nadzorowany przez doświadczonego w tym zakresie specjalistę;
3) którzy zdali egzamin przed komisją powołaną przez ministra właściwego do spraw zdrowia.
2. Przewodniczącym komisji, o której mowa w ust. 1 pkt 3, jest krajowy konsultant w dziedzinie radiologii i diagnostyki obrazowej.
3. Badania diagnostyczne i lecznicze wykonywane przy użyciu produktów radiofarmaceutycznych są wykonywane wyłącznie przez lekarzy posiadających specjalizację z medycyny nuklearnej lub pod ich nadzorem.
4. Badania rentgenodiagnostyczne są wykonywane przez lekarzy posiadających specjalizację z dziedziny radiologii lub techników elektroradiologii. Technicy elektroradiologii nie mogą wykonywać badań dynamicznych.
5. Lekarze, o których mowa w ust. 1–4, odbywają szkolenie specjalistyczne w dziedzinie ochrony radiologicznej, zakończone egzaminem przed komisją powołaną przez ministra właściwego do spraw zdrowia oraz potwierdzone certyfikatem.
6. Szkolenie, o którym mowa w ust. 5, powtarza się co 5 lat.
1) oczekiwane korzyści poznawcze lub lecznicze przewyższają niepożądane skutki napromienienia dla osób badanych lub leczonych;
2) będzie w nim brać udział jak najmniejsza liczba osób, przy możliwie małych dawkach promieniowania lub małych aktywnościach produktów radiofarmaceutycznych, zapewniających uzyskanie wyników na założonym poziomie prawdopodobieństwa znamienności statystycznej. Wartości dawek lub aktywności określane są przez pomiar i spełniają warunek optymalizacji ochrony przed promieniowaniem.
2. Osoby uczestniczące w eksperymencie, o którym mowa w ust. 1, przed przystąpieniem do niego:
1) są pisemnie informowane o ryzyku związanym z napromienieniem;
2) wyrażają zgodę na piśmie na uczestnictwo w eksperymencie.
3. Dawki, o których mowa w ust. 1 pkt 2, zależą od rodzaju i wielkości potencjalnych korzyści, o których mowa w ust. 4.
4. Wymaganą zależność między potencjalną korzyścią badania naukowego na ochotnikach przy użyciu produktów radiofarmaceutycznych lub promieniowania rentgenowskiego a wielkością ryzyka i dawką efektywną wyrażoną w milisiwertach (mSv) określa załącznik nr 3 do rozporządzenia.
2. Osoby, o których mowa w ust. 1, otrzymują szczegółową instrukcję postępowania opracowaną przez lekarza prowadzącego leczenie, po zasięgnięciu opinii krajowego konsultanta w dziedzinie terapii onkologicznej lub medycyny nuklearnej.
2. Dokumentacja systemu, o którym mowa w ust. 1, zawiera:
1) księgę jakości opracowaną zgodnie z normami PN-EN-ISO/IEC;
2) ogólne procedury opracowane zgodnie z normami, o których mowa w pkt 1;
3) opisy procedur postępowania diagnostycznego lub terapeutycznego mających zastosowanie w danym zakładzie opieki zdrowotnej, opracowane zgodnie z zaleceniami konsultantów krajowych w dziedzinie radioterapii onkologicznej, radiologii i diagnostyki obrazowej oraz medycyny nuklearnej;
4) instrukcje obsługi urządzeń radiologicznych;
5) zapisy dotyczące wyników przeprowadzanych wstępnych i okresowych testów urządzeń radiologicznych i urządzeń pomocniczych;
6) zapisy dotyczące kwalifikacji i szkoleń personelu;
7) zapisy dotyczące analizy odrzuconych wyników badań oraz podjętych działań korygujących i naprawczych;
8) zapisy dotyczące okresowych przeglądów systemu;
9) standardy opisów wyników badań.
Rozdział 2
Rentgenodiagnostyka
§ 12.
1) w radiografii planarnej stosuje się parametry ekspozycji decydujące o jakości i natężeniu promieniowania jonizującego właściwe dla fizycznej budowy pacjenta oraz rodzaju wykonywanego badania;
2) w radiografii planarnej liczba projekcji i czas ekspozycji oraz rozmiary wiązki promieniowania padającej na ciało pacjenta ogranicza się do wartości niezbędnych dla uzyskania żądanej informacji diagnostycznej;
3) stosuje się osłony osobiste chroniące przed promieniowaniem części ciała i narządy pacjenta niebędące przedmiotem badania, a znajdujące się w wiązce pierwotnej promieniowania, jeżeli nie umniejsza to diagnostycznych wartości wyniku badania;
4) stosuje się materiały i urządzenia zmniejszające do minimum narażenie na promieniowanie jonizujące i zapewniające uzyskanie obrazu o wartości diagnostycznej;
5) stosuje się wyłącznie automatyczne wywoływanie i utrwalanie błon rentgenowskich, z wyjątkiem błon stosowanych w stomatologii.
2. Podczas dokonywania diagnostycznych badań za pomocą rentgenowskiego tomografu komputerowego należy:
1) ograniczyć do niezbędnego minimum wielkość części ciała pacjenta będącej przedmiotem badania;
2) ograniczyć wartości iloczynu prądu i czasu ekspozycji wyrażonego w miliamperosekundach (mAs) do najmniejszych wartości pozwalających na uzyskanie obrazu o optymalnych cechach diagnostycznych;
3) dostosować parametry skanowania do rozmiarów ciała pacjenta;
4) w technice spiralnej z istniejących danych rekonstruować obraz warstw pośrednich zamiast wykonywania dodatkowych obrazów;
5) ograniczyć w technice spiralnej stosunek grubości warstwy badanej do skoku spirali do wartości nie większych od jedności;
6) zapewnić w technice stacjonarnej niezachodzenie na siebie warstw (przesunięcie stołu między kolejnymi warstwami jest nie mniejsze niż grubość warstwy);
7) stosować osłony osobiste na tarczycę, piersi, soczewki oczu i gonady, jeżeli leżą one bliżej niż 10 cm poza obszarem badanym, w szczególności u osób poniżej 16 roku życia;
8) stosować parametry ekspozycji dla dzieci uwzględniające wielkość ciała;
9) stosować rotację wiązki ograniczoną do kąta mniejszego od 360°, jeżeli jest to technicznie możliwe;
10) dokonać odpowiedniego wyboru parametrów rekonstrukcji obrazu;
11) stosować zmienną filtrację modyfikującą system akwizycji tomografu komputerowego, w przypadku gdy jest to możliwe;
12) rejestrować parametry ekspozycji.
3. Podczas diagnostycznych badań za pomocą mammografu należy;
1) używać urządzeń radiologicznych przeznaczonych wyłącznie do badań mammograficznych, których parametry odpowiadają normom serii EN 60601;
2) prowadzić systematyczną, cotygodniową kontrolę obrazu fantomu referencyjnego;
3) używać filtru rodowego w przypadku piersi o znacznych rozmiarach (powyżej 6 cm po kompresji) i dużej zawartości tkanki gruczołowej, jeżeli mammograf posiada możliwość wyboru rodzaju filtracji (Mo/Rh) z pulpitu sterowniczego;
4) ograniczyć do niezbędnego minimum stosowanie geometrycznego powiększenia obrazu;
5) stosować osłony osobiste ochraniające przed promieniowaniem jamę brzuszną, w szczególności w przypadku pacjentek w wieku rozrodczym;
6) stosować aparaty o ognisku nie większym niż 0,3 mm do mammograficznych badań przesiewowych dla odległości pomiędzy ogniskiem a detektorem równej 60 cm;
7) używać do oceny obrazów mammograficznych negatoskopów, o luminancji wyrażonej w kandelach na m2 większej niż 3 000 cd/m2.
4. W badaniach, o których mowa w ust. 1–3, należy stosować podstawowe parametry techniczne określone w załączniku nr 4 do rozporządzenia.
2. Testom wewnętrznej kontroli parametrów podlegają w szczególności:
1) parametry pracy lampy rentgenowskiej;
2) geometria wiązki promieniowania rentgenowskiego;
3) detektor obrazu;
4) systemy automatyki aparatu;
5) procesy fotochemiczne.
3. Sposób wykonywania wewnętrznej kontroli parametrów musi być zgodny z wymaganiami określonymi w normach, których wykaz ustala minister właściwy do spraw zdrowia, po zasięgnięciu opinii właściwego konsultanta krajowego.
4. Szczegółowy zakres i częstość testów, o których mowa w ust. 2, oraz dopuszczalne odchylenia badanych parametrów określa załącznik nr 5 do rozporządzenia.
5. Jeżeli zakres wewnętrznej kontroli parametrów wykracza poza możliwości techniczne zakładu opieki zdrowotnej, testy, o których mowa w ust. 2, wykonują jednostki organizacyjne posiadające notyfikację ministra właściwego do spraw zdrowia.
6. Nadzór wewnętrzny nad aparaturą rentgenodiagnostyczną sprawuje fizyk medyczny lub osoba posiadająca odpowiednie kwalifikacje.
7. Audyt kliniczny zewnętrzny sprawowany jest:
1) w zakresie stosowanych procedur i uzyskiwanych wyników – przez audytorów posiadających stosowne notyfikacje;
2) w zakresie narażenia pacjentów i parametrów technicznych aparatury rentgenodiagnostycznej – przez certyfikowane i notyfikowane w tym zakresie jednostki Państwowej Inspekcji Sanitarnej lub inne jednostki.
8. Zakres parametrów technicznych, o których mowa w ust. 7 pkt 2, oraz częstotliwość ich kontroli i tolerancję określa załącznik nr 6 do rozporządzenia.
2. Wniosek, o którym mowa w ust. 1:
1) zawiera szczegółowy opis systemu zarządzania jakością w zakładach opieki zdrowotnej, które zostały wyznaczone do prowadzenia badań przesiewowych;
2) określa cel i uzasadnienie badania;
3) wykazuje, że:
a) korzyści zdrowotne związane z badaniem przesiewowym przewyższają znacznie możliwe szkodliwe następstwa badania,
b) nie ma innych metod rozpoznawczych o podobnej skuteczności, obciążonych mniejszym ryzykiem.
3. Przeprowadzanie badań przesiewowych podlega okresowej kontroli w zakresie jakości wykonywania tych badań oraz ich wyników.
4. Kontrolę, o której mowa w ust. 3, przeprowadza zespół powoływany przez ministra właściwego do spraw zdrowia.
5. Zespół, o którym mowa w ust. 4, po każdej przeprowadzonej kontroli przedstawia ministrowi właściwemu do spraw zdrowia wniosek dotyczący kontynuowania lub zaprzestania badań przesiewowych.
6. Kontrola wyników badań przesiewowych obejmuje okresy nie dłuższe niż 3 lata, licząc od dnia wydania zgody, o której mowa w ust. 1.
2. Zabrania się wykonywania badań fotofluorograficznych u osób poniżej 16 roku życia.
1) używania wyłącznie aparatury rentgenodiagnostycznej wyposażonej w co najmniej sześciopulsowe generatory, z wyłączeniem badań stomatologicznych;
2) unieruchamiania niemowląt lub małych dzieci przy użyciu bobiksu lub innego urządzenia spełniającego tę funkcję;
3) stosowania osłon na gonady, a w przypadku tomografii klatki piersiowej u dziewcząt – także na gruczoły piersiowe, gdy w trakcie badania mogą znaleźć się w pobliżu pierwotnej wiązki promieniowania, jeżeli nie uniemożliwi to poprawnego wykonania badania.
2. Optymalne parametry techniczne badań rentgenodiagnostycznych z zakresu radiologii pediatrycznej określa tabela B załącznika nr 4 do rozporządzenia.
2. Badania, o których mowa w ust. 1, należy wykonywać w sposób zapewniający maksymalną ochronę płodu przed ekspozycją na promieniowanie, poprzez wybór właściwej techniki badania oraz stosowanie właściwych osłon na okolicę brzucha i miednicy.
3. Przepisów ust. 2 nie stosuje się w przypadku wykonywania badań rentgenodiagnostycznych koniecznych dla ratowania życia pacjentki.
2. W uzasadnionych przypadkach, w szczególności gdy z uwagi na stan pacjenta nie jest możliwe przeniesienie go do innego pomieszczenia, badanie lub zabieg, o których mowa w ust. 1, dokonywane są w pomieszczeniu, w którym się pacjent znajduje, przy czym:
1) wiązkę pierwotną promieniowania kierować należy wyłącznie na chorego;
2) inni chorzy, o ile jest to możliwe, opuszczają to pomieszczenie na czas badania lub zabiegu.
1) ukończyła 18 lat;
2) nie jest w ciąży;
3) wyposażona została w fartuch i rękawice ochronne z gumy ołowianej;
4) poinstruowana została o sposobie postępowania i ryzyku radiacyjnym.
2. Czynności, o których mowa w ust. 1, w warunkach ambulatoryjnych wykonywać może również członek rodziny pacjenta lub jego opiekun spełniający warunki określone w ust. 1.
3. Osoby, o których mowa w ust. 1, powinny być często zmieniane.
2. Aparatura, o której mowa w ust. 1, jest wyposażona w dobrze widoczny dla operatora rejestrator dawki i czasu ekspozycji.
3. Aparatura, o której mowa w ust. 1, jest poddawana systematycznej kontroli parametrów w zakresie funkcji i sprawności technicznej zgodnie z parametrami, o których mowa w ust. 4.
4. Zakres i częstotliwość wykonywania kontroli oraz tolerancję parametrów technicznych aparatury określa załącznik nr 6 do rozporządzenia.
2. Wykonywanie zabiegów zgodnie z procedurami, o których mowa w ust. 1, wymaga ponadto:
1) stosowania jak najkrótszego czasu emisji promieniowania jonizującego niezbędnego dla prawidłowego wykonania badania;
2) unikania trybu pracy aparatury rentgenowskiej w reżimie wysokiej mocy dawki;
3) właściwego doboru parametrów pracy lampy;
4) stosowania możliwie największej odległości lampy od pacjenta;
5) stosowania możliwie najbliższego położenia wzmacniacza obrazu względem ciała pacjenta;
6) ograniczenia do minimum stosowania geometrycznego powiększenia obrazu;
7) zmieniania położenia miejsca wejścia wiązki pierwotnej promieniowania;
8) ograniczenia do minimum liczby rejestrowanych obrazów;
9) stosowania kontroli indywidualnych dawek promieniowania jonizującego otrzymywanych przez personel na całe ciało oraz na skórę dłoni.
2. Dalsze postępowanie lekarskie zależne jest od wielkości dawki promieniowania obliczonej zgodnie z ust. 1 oraz szacunku ryzyka poważnych uszkodzeń zarodka lub płodu.
2. W stosunku do pacjenta, o którym mowa w ust. 1, w przypadku gdy jest to konieczne, podejmuje się leczenie dermatologiczne.
3. Jeżeli pacjent w wyniku zabiegu z zakresu radiologii interwencyjnej, wykonywanego według obowiązujących procedur, a mogącego wymagać powtórzenia, otrzymał dawkę sumaryczną na skórę przekraczającą 1 Gy, dokumentacja wyników badań i informacja o dawce jest przekazywana lekarzowi prowadzącemu.
4. Zabiegi wykonywane według procedur, o których mowa w ust. 3, wymagają rejestracji i rozpoznawania ekspozycji pacjenta oraz przekazywania tych informacji innym lekarzom i placówkom uczestniczącym w jego leczeniu.
2. Audyt zewnętrzny w zakładach, o których mowa w ust. 1, przeprowadza komisja powołana przez konsultanta krajowego w dziedzinie radiologii i diagnostyki obrazowej co najmniej raz na 3 lata, a także po każdym incydencie prowadzącym do uchwytnego uszkodzenia popromiennego u pacjenta.
3. Zakład, o którym mowa w ust. 1, zgłasza niezwłocznie ministrowi właściwemu do spraw zdrowia wystąpienie incydentów, o których mowa w ust. 2.
Rozdział 3
Medycyna nuklearna
§ 26.
1) przyjmowanie i przechowywanie źródeł promieniotwórczych;
2) prowadzenie prac związanych z preparatyką i dozowaniem produktów radiofarmaceutycznych;
3) hospitalizowanie pacjentów poddawanych onkologicznej terapii jodem-131, jeżeli taka terapia jest prowadzona;
4) zbieranie i przechowywanie skażonej pościeli, bielizny i odpadów promieniotwórczych;
5) kontrolę dozymetryczną, usuwanie skażeń i przechowywanie odzieży (śluza, punkt dozymetryczny, szatnia), wyposażone w kabiny z prysznicem i umywalkami;
6) aparaturę diagnostyczną;
7) laboratorium fotochemiczne, jeżeli stosuje się chemiczną obróbkę błon fotograficznych;
8) gabinet lekarski;
9) poczekalnię z możliwością oddzielenia przestrzeni zajmowanej przez pacjentów przed i po podaniu produktów radiofarmaceutycznych;
10) węzeł sanitarny.
2. Wyjścia z oddziału, w którym do terapii onkologicznej stosuje się otwarte źródła promieniotwórcze, są monitorowane, w szczególności za pomocą bramki dozymetrycznej z sygnałem dźwiękowym.
1) wykonywania badań diagnostycznych i przeprowadzania leczenia wyłącznie na podstawie pisemnych procedur zgodnie z opinią właściwego konsultanta krajowego;
2) wykonywania wszelkich czynności związanych z przygotowaniem produktów radiofarmaceutycznych przez znakowanie gotowych zestawów lub podzielenie większych porcji gotowych produktów radiofarmaceutycznych do podania pacjentom, wyłącznie w przeznaczonych do tego celu pomieszczeniach wyposażonych w wyciągi radiochemiczne i zapewniających zachowanie jałowości w procesie znakowania;
3) w przypadku gdy w zakładzie medycyny nuklearnej znakuje się radionuklidem pobrany od pacjenta materiał biologiczny oraz w każdym przypadku wytwarzania produktu radiofarmaceutycznego, wydzielone do tego celu pomieszczenia i tryb pracy zapewniają utrzymanie stopnia czystości bakteriologicznej klasy A w rozumieniu przepisów w sprawie wymagań Dobrej Praktyki Wytwarzania;
4) stosowania osłon przed promieniowaniem jonizującym właściwych dla rodzaju źródeł i zakresu prowadzonych prac, w tym używania strzykawek wyposażonych w osłony pochłaniające promieniowanie gamma i beta;
5) niedozwolona jest manipulacja źródłami o aktywności wyrażonej w megabekerelach większej niż 0,1 MBq nieosłoniętymi osłoną o grubości co najmniej równoważnej 3 mm Pb w sposób umożliwiający ich dotykowy kontakt ze skórą dłoni i palców, również osłoniętych rękawicą zapobiegającą skażeniom powierzchniowym;
6) przy podawaniu pacjentom produktów radiofarmaceutycznych w celach diagnostycznych należy stosować metody postępowania ograniczające odkładanie się znacznika promieniotwórczego w narządach niepodlegających badaniu oraz – jeżeli jest to możliwe – przyspieszające wydalanie znacznika z organizmu pacjenta;
7) każdorazowe podanie pacjentowi produktu radiofarmaceutycznego wymaga uprzedniego zmierzenia jego aktywności, tak aby pacjent otrzymał ilość (aktywność) przepisaną przez lekarza nadzorującego badanie;
8) podawanie produktu radiofarmaceutycznego dorosłym pacjentom uwzględnia – w przypadkach, w których jest to uzasadnione, ciężar lub powierzchnię ciała, a w przypadku osób do 16 roku życia – ciężar ciała lub wiek; sposoby obliczania aktywności opiniuje konsultant krajowy w dziedzinie medycyny nuklearnej;
9) informowania na piśmie pacjenta poddawanego terapii radioizotopowej o właściwym zachowaniu się w stosunku do najbliższego otoczenia zgodnie z opinią konsultanta krajowego w dziedzinie medycyny nuklearnej.
2. W przypadkach, o których mowa w ust. 1, należy:
1) ograniczyć aktywności produktów radiofarmaceutycznych do najmniejszej wartości umożliwiającej badanie;
2) zwiększyć podaż płynów badanej;
3) pouczyć badaną o konieczności częstego oddawania moczu.
3. Stosowanie do celów diagnostycznych i leczniczych jodków znakowanych 131I i 125I u kobiet w ciąży po 8 tygodniach od zapłodnienia nie jest dopuszczalne. Dotyczy to również leczenia przeciwbólowego przy użyciu osteotropowych produktów radiofarmaceutycznych w dowolnym okresie ciąży.
4. W przypadku konieczności wykonania badania lub leczenia przy użyciu produktu radiofarmaceutycznego u kobiety karmiącej, lekarz wykonujący lub nadzorujący badanie jest obowiązany poinformować pacjentkę o konieczności przerwania karmienia piersią lub okresowego zaprzestania karmienia, z podaniem długości tego okresu.
5. Okresy zaprzestania karmienia piersią niemowląt po podaniu produktów radiofarmaceutycznych dla celów diagnostycznych określa załącznik nr 7 do rozporządzenia.
2. Jeżeli podana jednorazowa aktywność przekracza wartość określoną w ust. 1, pacjent może być zwolniony ze szpitala po spadku aktywności w ciele poniżej tej wartości.
3. Przy podejmowaniu decyzji o zwolnieniu z leczenia szpitalnego otwartymi źródłami jodu-131 uwzględnić należy każdorazowo warunki mieszkaniowe i rodzinne pacjenta oraz możliwości przestrzegania przez niego ograniczeń warunkujących zmniejszenie ryzyka radiacyjnego dla osób z otoczenia, tak aby dawki efektywne dla tych osób nie przekroczyły wartości, o których mowa w ust. 4. Przepisu nie stosuje się do osób, o których mowa w § 8.
4. Limity użytkowe dawek dla planowania ochrony przed promieniowaniem rodziny pacjenta leczonego otwartymi źródłami jodu-131 oraz osób postronnych określa załącznik nr 8 do rozporządzenia.
5. Przy podejmowaniu decyzji, o której mowa w ust. 3, należy uwzględnić opinie konsultanta krajowego w dziedzinie medycyny nuklearnej.
2. Minimalny zakres i częstotliwość testów, o których mowa w ust. 1, określa załącznik nr 9 do rozporządzenia.
2. W przypadku zakładu kierowanego przez konsultanta, o którym mowa w ust. 1, audyt kliniczny zewnętrzny prowadzony jest przez osobę wyznaczoną przez konsultanta krajowego w dziedzinie medycyny nuklearnej.
3. Audyt kliniczny zewnętrzny jest wykonywany nie rzadziej niż co 3 lata, a jego wyniki przekazywane konsultantowi krajowemu w dziedzinie medycyny nuklearnej.
2. Częstotliwość kontroli, o której mowa w ust. 1, uzależnia się od stopnia zagrożenia skażeniami wewnętrznymi jodem-131.
3. W zakładach, w których stosuje się znakowanie produktów radiofarmaceutycznych izotopem 99mTc uzyskiwanym z generatora, należy oznaczać indywidualne dawki promieniowania dla skóry dłoni osób przeprowadzających procedury znakowania.
Rozdział 4
Radioterapia
§ 35.
1) właściwą strukturę organizacyjną i wyposażenie zakładu;
2) właściwy dobór, liczbę i kwalifikacje personelu;
3) przestrzeganie ustalonego regulaminu pracy zakładu, procedur dotyczących jakości i kontroli tej jakości, w tym uczestnictwa w audytach wewnętrznych i zewnętrznych.
2. Wymagania dotyczące pomieszczeń i wyposażenia zakładu teleradioterapii określa załącznik nr 10 do rozporządzenia.
3. Wymagania dotyczące wyposażenia zakładu brachyterapii określa załącznik nr 11 do rozporządzenia.
2. Niedopuszczalne jest stosowanie aparatów ortowoltowych do celów radioterapii głębokiej.
2. Rejestr eksploatacji zawiera w szczególności informacje o:
1) awariach;
2) przeprowadzonych konserwacjach i naprawach;
3) dokonanych kontrolach;
4) innych zdarzeniach mogących mieć wpływ na pracę aparatu terapeutycznego i symulatora.
3. Wpisu do rejestru eksploatacji dokonywać należy w sposób czytelny, opatrując go podpisem i datą.
2. Urządzenia dozymetryczne oraz urządzenia wspomagające planowanie i realizację leczenia wykorzystywane w zakładach radioterapii podlegają okresowym wewnętrznym testom kontrolnym.
3. Szczegółowe zasady, zakres oraz częstotliwość przeprowadzania kontroli, o których mowa w ust. 1 i 2, określa załącznik nr 12 do rozporządzenia.
4. Dokumenty z przeprowadzonych kontroli, o których mowa w ust. 1 i 2, przechowuje się przez co najmniej 5 lat od dnia zakończenia eksploatacji aparatu lub urządzenia.
2. W zakładzie radioterapii, w którym prowadzone jest radykalne leczenie przeciwnowotworowe, liczba zatrudnionych lekarzy ze specjalnością z radioterapii onkologicznej i techników radioterapii zapewnia:
1) uczestnictwo lekarza w czasie pierwszego napromieniania pacjenta;
2) obecność dwóch techników podczas napromieniania pacjenta z użyciem zewnętrznych wiązek promieniowania megawoltowego.
3. Przy zakładzie radioterapii działa pracownia (zakład) fizyki medycznej.
4. Pracownią (zakładem) fizyki medycznej lub pracownią (zakładem) planowania leczenia kieruje fizyk medyczny, który jest odpowiedzialny za planowanie i sprawdzanie fizycznych oraz technicznych parametrów napromieniania.
5. W zakładzie radioterapii wdraża się i dokumentuje procedurę podwójnego niezależnego sprawdzania:
1) wyznaczonej objętości tarczowej i technik napromieniania;
2) obliczonego czasu napromienienia lub obliczonej ekspozycji wyrażonej w jednostkach monitorowych, dla każdego obszaru napromieniania;
3) fizycznych i technicznych parametrów ustawienia aparatu terapeutycznego oraz parametrów technicznych stołu terapeutycznego.
6. Wymagania procedur systemu zarządzania i kontroli jakości radioterapii określa załącznik nr 13 do rozporządzenia.
2. Odstępstwa od protokołu terapeutycznego są uzasadniane w dokumentacji medycznej.
2. Plan leczenia, o którym mowa w ust. 1, zatwierdza co najmniej dwóch lekarzy specjalistów w dziedzinie radioterapii onkologicznej.
3. W przypadku brachyterapii, dane, o których mowa w ust. 1, obejmują w szczególności:
1) wyznaczenie kolejności prowadnic przeznaczonych do poruszania się źródeł i ich oznaczenie;
2) wyznaczenie na podstawie zdjęć lokalizacyjnych przestrzennych współrzędnych źródeł promieniotwórczych.
4. Odstąpienie od wykonania zdjęć, o których mowa w ust. 3 pkt 2, uzasadnione jest w przypadku, gdy układ aplikatorów w sposób jednoznaczny określa współrzędne źródeł.
5. W przypadku teleradioterapii dane, o których mowa w ust. 1, obejmują symulację zaplanowanych wiązek. Dokonanie symulacji utrwala się zapisem w postaci zdjęcia rentgenowskiego lub zapisu cyfrowego. Odstąpienie od dokonania symulacji uzasadniać mogą jedynie względy medyczne każdorazowo odnotowywane w dokumentacji medycznej pacjenta.
6. W odniesieniu do trójwymiarowego planowania leczenia wymagane są:
1) seria zdjęć tomograficznych w odstępach nie większych niż 8 mm;
2) trójwymiarowe odtworzenie objętości tarczowej i narządów krytycznych;
3) udokumentowanie planu leczenia w formie histogramu rozkładu dawki w objętości tarczowej.
7. Lekarz prowadzący leczenie na bieżąco wpisuje do karty napromieniania dane zawarte w planie leczenia lub podpisem potwierdza wpis technika radioterapii.
2. Kontrola karty napromieniania jest udokumentowana. Szczegółowej podwójnej kontroli podlegają:
1) prawidłowość obliczeń czasów napromieniania dla aparatów ze źródłem promieniotwórczym;
2) prawidłowość obliczeń dawki monitorowej dla akceleratorów;
3) dawki sumaryczne otrzymywane przez pacjenta;
4) zgodność wpisów w karcie napromieniania z opisem planu leczenia.
3. W brachyterapii z zastosowaniem urządzeń ze zdalnie sterowanymi źródłami promieniotwórczymi kontrola planu leczenia i karty napromieniania obejmuje:
1) ocenę prawidłowości obliczeń czasu postoju źródeł w zaplanowanych punktach;
2) spełnienie wymagań określonych w ust. 2 pkt 3 i 4.
4. Kontrola czasu i dawki napromieniania jest dokonywana najpóźniej po pierwszym napromienianiu. W przypadku gdy pacjent jest napromieniony jednorazowo, kontrola dokonywana jest przed ekspozycją.
5. Przeprowadzający kontrolę potwierdza jej dokonanie podpisem i datą.
1) dane jednoznacznie identyfikujące pacjenta oraz rozpoznanie lekarskie;
2) nazwisko lekarza prowadzącego, a w przypadku jego czasowej nieobecności – nazwisko lekarza zastępującego, a także lekarza nadzorującego;
3) czytelnie i jednoznacznie sformułowane, opatrzone podpisem lekarza, dyspozycje realizacji napromieniania z uwzględnieniem:
a) parametrów ekspozycji terapeutycznych pacjenta na stole terapeutycznym oraz punktów topograficznych dla centratorów pozycjonowania pacjenta,
b) wartości dawki frakcyjnej w obszarze tarczowym dla każdego pola napromieniania (lub wartości łącznej będącej sumą przyczynków z wszystkich pól napromieniania),
c) przedziału czasowego między kolejnymi frakcjami,
d) wartości dawki całkowitej i sposobu zapisu jej akumulacji,
e) dla zdefiniowanych i wybranych narządów krytycznych – analogiczne wartości parametrów określonych w lit. b–d dotyczą maksymalnej dawki,
f) zdefiniowanych użytych modyfikatorów (osłony, filtry, kompensatory) i przyporządkowanie ich odpowiednim polom napromieniania wraz z opisem ich użycia.
2. W trakcie realizacji radioterapii technik potwierdza podpisem zgodność parametrów zapisanych ze zrealizowanymi, w szczególności jednostek monitorowych (czas ekspozycji).
1) uczestniczenia lekarza oraz fizyka medycznego na wniosek lekarza prowadzącego, potwierdzonego ich podpisem, w przygotowaniu pacjenta do pierwszego napromieniania;
2) uczestniczenia co najmniej dwóch techników radioterapii w przygotowaniu pacjenta do napromieniania;
3) stałej obserwacji pacjenta przez technika radioterapii podczas napromieniania;
4) udziału fizyka medycznego w czasie napromieniania – na wniosek lekarza lub technika.
2. Aparat terapeutyczny jest okresowo wyłączany z eksploatacji w celu konserwacji, kontroli parametrów technicznych oraz kalibracji i dozymetrii generowanego promieniowania zgodnie z przyjętym wewnętrznym harmonogramem jego pracy.
3. Harmonogram, o którym mowa w ust. 2, ustala kierownik zakładu radioterapii w porozumieniu z fizykiem medycznym, uwzględniając, że w okresie godziny może być leczonych radykalnie nie więcej niż 5 pacjentów.
4. Lekarz specjalista z dziedziny radioterapii planujący i realizujący leczenie z wykorzystaniem promieniowania jonizującego jest odpowiedzialny za prawidłowość proponowanego leczenia i jego skutki kliniczne.
1) przygotowania pacjenta do leczenia, planowania i realizacji napromieniania przez lekarza specjalistę z dziedziny radioterapii oraz jego uczestnictwa w rozpoczęciu napromieniania;
2) stałej obserwacji pacjenta w czasie napromieniania;
3) umieszczania pacjenta z wprowadzonymi na stałe źródłami promieniotwórczymi w odizolowanym pomieszczeniu do czasu zmniejszenia mocy dawki ekspozycyjnej do wartości dopuszczalnej dla osób postronnych;
4) w przypadku bezpośrednich aplikacji źródeł promieniotwórczych – stosowania osłon i narzędzi pozwalających zmniejszyć do minimum narażenie personelu na promieniowanie pod warunkiem, że nie utrudni to implantacji;
5) zapewnienia możliwości monitorowania wyjścia pracownika z obszaru kontrolowanego, w szczególności przez bramkę dozymetryczną z sygnałem dźwiękowym, w zakładach brachyterapii stosujących ręczne aplikacje źródeł promieniotwórczych;
6) wykonywania zdjęć sprawdzających położenie zaaplikowanych źródeł promieniotwórczych bezpośrednio w pomieszczeniu, w którym dokonuje się aplikacji;
7) zabezpieczenia źródeł promieniotwórczych na czas aplikacji przed przypadkowym przemieszczeniem;
8) okresowego sprawdzania położenia źródeł promieniotwórczych – w przypadku długotrwałych aplikacji;
9) porównania po skończonym leczeniu liczby źródeł promieniotwórczych użytych do aplikacji z liczbą źródeł wyjętych oraz dodatkowej kontroli pacjenta za pomocą detektora promieniowania;
10) przygotowania indywidualnych aplikatorów oraz stosowania technik sterylizacji wykluczających możliwość uszkodzenia źródeł promieniotwórczych;
11) udziału fizyka medycznego w procedurach wymienionych w pkt 1 i pkt 6–10.
1) lokalizacji guza w stosunku do płodu;
2) zastosowania osłon chroniących płód w przypadku, gdy odległość i położenie guza to umożliwiają;
3) ustalenia ryzyka dla matki wynikającego z leczenia innego niż radioterapia;
4) obliczenia dawki dla zarodka lub płodu, będącej wynikiem podjętej terapii;
5) ustalenia prawdopodobieństwa uszkodzenia płodu w zależności od okresu ciąży, w którym nastąpiłoby leczenie.
2. Jeżeli przeprowadzone oceny wskazują na wysokie prawdopodobieństwo powstania ciężkiego uszkodzenia płodu polegającego na powstaniu wad rozwojowych poszczególnych narządów, ciężkiego niedorozwoju umysłowego, wysokiego prawdopodobieństwa indukcji nowotworu, który może ujawnić się w okresie pierwszych 20 lat życia dziecka, kobieta ciężarna jest o tym informowana przez konsylium lekarskie, celem podjęcia decyzji o dalszym postępowaniu.
3. Dla uniknięcia niezamierzonego uszkodzenia zarodka lub płodu w wyniku radioterapii okolicy brzucha i miednicy, w przypadku nierozpoznawalnej ciąży, można podjąć radioterapię u kobiet w okresie płodności wyłącznie po uzyskaniu negatywnego testu ciążowego, przeprowadzonego u pacjentki przed podjęciem decyzji o leczeniu.
4. Od wykonania testu, o którym mowa w ust. 3, można odstąpić, jeżeli istnieją bezpośrednie okoliczności świadczące o niemożliwości zajścia pacjentki w ciążę.
Rozdział 5
Nieszczęśliwe wypadki związane ze stosowaniem promieniowania jonizującego w radioterapii oraz szczegółowe zasady zapobiegania tym wypadkom
§ 47.
2. W przypadku, o którym mowa w ust. 1, technik radioterapii obsługujący aparat terapeutyczny obowiązany jest zgłosić awarię osobie odpowiedzialnej za stan i sprawność aparatury w zakładzie.
3. Technik radioterapii może użytkować aparat terapeutyczny, który miał awarię, po otrzymaniu protokołu dopuszczenia aparatu do dalszej pracy podpisanego przez kierownika zakładu.
4. Osobę odpowiedzialną za stan i sprawność aparatury w zakładzie wyznacza dyrektor zakładu opieki zdrowotnej lub osoba przez niego upoważniona.
5. Kierownik zakładu radioterapii, brachyterapii lub medycyny nuklearnej jest obowiązany prowadzić rejestr i dokumentację błędów technicznych i dozymetrycznych oraz wszelkich niezgodności między parametrami i wskaźnikami zapisanymi w karcie napromienienia a parametrami i wskaźnikami w trakcie realizacji napromieniania, prowadzących do wystąpienia istotnego błędu dozymetrycznego.
6. Kierownik zakładu, o którym mowa w ust. 5, obowiązany jest:
1) wyjaśnić przyczynę i uwarunkowania stwierdzonego błędu;
2) powiadomić bezpośredniego przełożonego o powstałym błędzie;
3) podjąć działania zmierzające do eliminacji przyczyn błędu.
2. Jeżeli pacjentowi podano pomyłkowo produkt radiofarmaceutyczny o aktywności wyższej niż przepisana – to należy, jeżeli jest to technicznie możliwe, wdrożyć postępowanie zmierzające do zmniejszenia wychwytu nuklidu w narządach i przyspieszenia jego wydalania z ustroju.
3. Konsultant krajowy w dziedzinie radioterapii lub medycyny nuklearnej nakazuje niezwłocznie przeprowadzenie audytu zewnętrznego celem wykrycia przyczyn i zapobieżenia w przyszłości zdarzeniom, o których mowa w ust. 1 i 2.
2. W przypadku gdy przyczyną wypadku w radioterapii była lub mogła być awaria aparatu terapeutycznego, kierownik zakładu:
1) wstrzymuje napromienianie terapeutyczne przy zastosowaniu tego urządzenia;
2) zabezpiecza urządzenie oraz pomieszczenie, w którym się ono znajduje, przed dostępem z zewnątrz.
3. W przypadku gdy wypadek w radioterapii powstał w wyniku błędu proceduralnego, kierownik zakładu radioterapii lub medycyny nuklearnej do czasu wyjaśnienia przyczyn wypadku zakazuje uczestniczenia w leczeniu pacjentów osobom biorącym udział w procesie leczenia pacjentów, którzy ulegli wypadkowi.
2. Komisja, o której mowa w ust. 1, przystępuje do wykonywania czynności wyjaśniających niezwłocznie i przygotowuje oraz przekazuje niezwłocznie raport ministrowi właściwemu do spraw zdrowia.
2. Audyt, o którym mowa w ust. 1, dokonywany jest na zlecenie ministra właściwego do spraw zdrowia przez powołany w tym celu zespół.
3. Przeprowadzenie audytu, o którym mowa w ust. 1, nie wyklucza przeprowadzenia kontroli wykonywanych na podstawie odrębnych przepisów.
1) spełnieniu wymagań określonych w § 7;
2) uzyskaniu pozytywnej opinii właściwego konsultanta krajowego w sprawie wprowadzenia tych technik lub sposobów frakcjonowania dawki promieniowania.
2. Dokonując oceny projektu zastosowania niekonwencjonalnej techniki, konsultant, o którym mowa w ust. 1 pkt 2, oraz komisja bioetyczna, o której mowa w § 7, rozpatrują w szczególności:
1) właściwość kwalifikacji pacjenta do proponowanej techniki leczenia;
2) jakość i stopień uzasadnienia podjęcia leczenia, w szczególności teoretyczne i eksperymentalne dane przedstawione przez autorów projektu;
3) prawdopodobieństwo negatywnego wyniku badania i wynikające z tego możliwe następstwa dla zdrowia pacjentów;
4) opinie ekspertów powołanych przez komisję bioetyczną.
3. Zapewnia się obiektywną ocenę skutków leczenia niekonwencjonalną techniką i obserwacje pacjentów przez okres niezbędny dla pełnej oceny rezultatów leczenia.
4. W celu uzyskania opinii, o której mowa w ust. 1 pkt 2, zakład, o którym mowa w ust. 1, powinien:
1) posiadać udokumentowane przeszkolenie w zakresie umiejętności praktycznych lekarzy specjalistów, fizyków medycznych i techników do stosowania konwencjonalnych i niekonwencjonalnych procedur;
2) udokumentować wdrożenie wszystkich wymaganych procedur kontroli jakości, o których mowa w § 39;
3) udokumentować zakres możliwości technicznych aparatury symulacyjnej i terapeutycznej do planowania i realizacji zadań, o których mowa w ust. 1.
Rozdział 6
Przepisy przejściowe i końcowe
§ 55.
2. Systemy zarządzania jakością, o których mowa w § 9, są certyfikowane, zgodnie z odrębnymi przepisami, w terminie do dnia 31 grudnia 2005 r.
2. W przypadku gdy aparatura, o której mowa w ust. 1, z przyczyn technicznych nie może zostać wyposażona w terminie do dnia 31 grudnia 2004 r. w urządzenia, o których mowa w § 21 ust. 2, powinna ona zostać wycofana z eksploatacji w terminie do dnia 31 grudnia 2005 r.
3. Testy, o których mowa w § 21 ust. 3, wdrażane są w terminie do dnia 31 grudnia 2003 r.
Minister Zdrowia: M. Łapiński
|
|
1) Minister Zdrowia kieruje działem administracji rządowej – zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 28 czerwca 2002 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. Nr 93, poz. 833).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 24 grudnia 2002 r. (poz. 2098)
Załącznik nr 1
POZIOMY REFERENCYJNE DAWEK DLA BADAŃ RENTGENODIAGNOSTYCZNYCH
A. Radiografia i mammografia
| Lp. | Rodzaj badania | Dawka(1) [mGy] |
| 1 | Radiografia klatki piersiowej |
|
|
| – projekcja AP | 0,3 |
|
| – projekcja LAT | 1,5 |
| 2 | Radiografia czaszki |
|
|
| – projekcja AP/PA | 5 |
|
| – projekcja LAT | 3 |
| 3 | Radiografia kręgosłupa lędźwiowego |
|
|
| – projekcja AP/PA | 10 |
|
| – projekcja LAT | 30 |
|
| – projekcja LAT stawu lędźwiowo-krzyżowego | 40 |
| 4 | Radiografia miednicy |
|
|
| – projekcja AP | 10 |
| 5 | Radiografia układu moczowego |
|
|
| – projekcja AP zwykłe zdjęcie lub przed podaniem |
|
|
| środka kontrastowego | 10 |
|
| – po podaniu środka kontrastowego | 10 |
| 6 | Mammografia(2) |
|
|
| – projekcja MLO | 10 |
|
| – projekcja CC | 10 |
(1) Poziomy referencyjne dawek rozumiane jako: wejściowa dawka powierzchniowa odnoszą się do standardowego pacjenta o wzroście 170 cm i masie 70 kg i są wyrażone jako dawka pochłonięta w powietrzu w punkcie przecięcia osi wiązki z powierzchnią ciała pacjenta.
(2) Wartości wejściowej dawki powierzchniowej odnoszą się do 5 cm ściśnięcia piersi dla standardowego pacjenta przy zdjęciu z wykorzystaniem kratki przeciwrozproszeniowej.
B. Radiologia pediatryczna
| Lp. | Rodzaj badania(1) | Dawka (2) [μGy] |
| 1 | Radiografia klatki piersiowej (poza noworodkami) |
|
| – projekcja PA/AP | 100 | |
| – projekcja LAT | 200 | |
| 2 | Radiografia klatki piersiowej noworodków | 80 |
| – projekcja AP | ||
| 3 | Radiografia czaszki |
|
| – projekcja PA/AP | 1500 | |
| – projekcja LAT | 1000 | |
| 4 | Radiografia miednicy |
|
| – niemowlęta | 200 | |
| – starsze dzieci | 900 | |
| 5 | Radiografia brzucha z użyciem wiązki poziomej lub pionowej | 1000 |
(1) Wartości poziomów referencyjnych dla pozostałych badań są obecnie nieokreślone.
(2) Poziomy referencyjne odnoszą się do standardowego pacjenta w wieku 5 lat (z wyłączeniem badań noworodków i niemowląt).
C. Tomografia komputerowa
| Lp. | Rodzaj badania | Dawka | |
| CTDI | DLP | ||
| 1 | Rutynowe badania głowy lub mózgu(2) | 60 | 1050 |
| 2 | Badanie twarzy i zatok(2) | 35 | 360 |
| 3 | Badanie urazów kręgów(3) | 70 | 460 |
| 4 | Rutynowe badania klatki piersiowej(3) | 30 | 650 |
| 5 | Wysokorozdzielcze badania płuc(3) | 35 | 280 |
| 6 | Rutynowe badanie brzucha lub jamy brzusznej(3) | 35 | 780 |
| 7 | Badanie wątroby i śledziony(3) | 35 | 900 |
| 8 | Rutynowe badania miednicy lub narządów miednicy(3) | 35 | 570 |
| 9 | Badanie kości miednicy lub obręczy biodrowej(3) | 25 | 520 |
(1) Referencyjne poziomy dawek określone na podstawie dawki pochłoniętej w powietrzu.
(2) Dane odnoszą, się do fantomu głowy (PMMA o średnicy 16 cm).
(3) Dane odnoszą się do fantomu ciała (PMMA o średnicy 32 cm).
Załącznik nr 2
POZIOMY REFERENCYJNE AKTYWNOŚCI PRODUKTÓW RADIOFARMACEUTYCZNYCH, PODAWANYCH DOROSŁYM PACJENTOM O TYPOWEJ BUDOWIE CIAŁA (CIĘŻAR – 70 KG, WZROST – 170 CM) W NAJCZĘSTSZYCH BADANIACH DIAGNOSTYCZNYCH
| Rodzaj badania | Radionuklid i produkt radiofarmaceutyczny | Aktywność na badanie (MBq) |
| 1 | 2 | 3 |
| Kościec – obrazowanie | 99mTc fosforany, fosfoniany | 750 |
| Szpik kostny – obrazowanie | 99mTc – koloidy | 400 |
| Perfuzja mózgu | 99mTc – HmPAO | 750 |
| 99mTc – ECD | 750 | |
| Cysternografia | 111In DTPA | 40 |
| Obrazowanie tarczycy | 99mTcO4– | 80 |
| 123I – jodki | 20 | |
| 131I – odki | 4 | |
| Poszukiwanie przerzutów raka tarczycy po ablacji gruczołu | 131I – jodki | 240 |
| Obrazowanie przytarczyc i gruczolaków tego narządu | 99mTcMIBi | 750 |
| Obrazowanie wentylacji płuc | 133 Xe – gaz w roztworze | 400 |
| 127 Xe – gaz w roztworze | 200 | |
| 99m Tc – DTPA – aerozol | 200 | |
| Planarne obrazowanie perfuzji płuc | 99mTc – mikrosfery | 100 |
| Tomograficzne obrazowanie perfuzji płuc | 99mTc – mikrosfery | 400 |
| Obrazowanie wątroby i śledziony | 99mTc – znakowane koloidy | 200 |
| Obrazowanie dynamiczne układu żółciowego | 99mTc – pochodne iminodwuoctanu | 200 |
| Obrazowanie śledziony zdenaturowanymi erytrocytami | 99mTc – erytrocyty zdenat. | 100 |
| Badanie pierwszego przejścia krwi przez krążenie płucne i serce | 99mTCO4–-– roztwór | 400 |
| 99mTc DTPA | 800 | |
| Obrazowanie puli krwi w lewej komorze i dynamika jej pracy (bramkowanie) | 99mTc – erytrocyty (znakowane in vivo) | 800 |
| Obrazowanie i perfuzja m. sercowego lewej komory | 99mTc – fosfoniany, izonitryle i równoważne 201 | 800 |
| 100 | ||
| Obrazowanie uchyłku Meckel’a | 99mTcO4– – roztwór | 400 |
| 1 | 2 | 3 |
| Krwawienie z przewodu pokarmowego – lokalizacja | 99mTc – erytrocyty i równoważne | 400 |
| Badanie przejścia pokarmu przez przełyk, badanie refluksu przełykowego | 99mTc – koloidy i związki niewchłanialne | 40 |
| Badanie opróżniania żołądka | niewchłanialne związki 994Tc | 40 |
| Statyczne obrazowanie nerek | 99mTc – DMSA | 200 |
| Dynamiczne badanie układu moczowego | 99mTc – DTPA | 200 |
| 99mTc – EC, MAG-3 | 100 | |
| 123I – o-hipuran | 20 | |
| Obrazowanie nadnerczy | 131I – metylocholesterol | 40 |
| Obrazowanie wybranych nowotworów i ropni | 67Ga – cytrunian | 400 |
| Obrazowanie wybranych nowotworów | 99mTc – analogi somatostatycznych | 800 |
| Obrazowanie guzów neuroektodermalnych | 123I – metajodobenzyloguanidyna | 400 |
| 131I – metajodobenzyloguanidyna | 40 | |
| Obrazowanie rozległości procesu nowotworowego wybranych guzów | 99mTc – MIBI | 1000 |
| Obrazowanie strażniczych węzłów chłonnych | 99mTc – koloidy | 80 |
| Obrazowanie ropni i ognisk zapalnych | 99mTc – znak. leukocyty | 800 |
| 99mTc – immunoglobulina | 400 | |
| Oznaczenie klirensu nerkowego kłębkowego | 994Tc DTPA | 40 |
| Oznaczenie efektywnego przepływu osocza przez nerki | 99mTc – EC | 40 |
| 123I – ortohipuran | 20 | |
| 131I – ortohipuran | 6 | |
| Szybkość oczyszczania osocza na drodze sekrecji kanalikowej | 99mTc MAG3 | 40 |
| Wątrobowy klirens 99mTc – HEPIDA | 99mTe HEPIDA | 40 |
Załącznik nr 3
WYMAGANA ZALEŻNOŚĆ MIĘDZY POTENCJALNĄ KORZYŚCIĄ BADANIA NAUKOWEGO NA OCHOTNIKACH PRZY UŻYCIU PRODUKTÓW RADIOFARMACEUTYCZNYCH LUB PROMIENIOWANIA RENTGENOWSKIEGO A WIELKOŚCIĄ RYZYKA l DAWKĄ EFEKTYWNĄ W mSv
| Poziom ryzyka radiacyjnego w ciągu całego życia (prawdopodobieństwo) | Dawka efektywna (mSv) | Rodzaj oczekiwanej potencjalnej korzyści z badania |
| znikomy (<10-6) | <0.1 | rozwój wiedzy |
| b. mały (~10-5) | 0.1 – 1.0 | rozwój wiedzy prowadzący do potencjalnych korzyści zdrowotnych |
| pośredni (~10-4) | 1 –10 | bezpośredni cel w postaci polepszenia leczenia lub zapobiegania chorobie |
| umiarkowany (>10-3) | >10 | znaczny, bezpośrednio związany z ratowaniem życia, zapobieganiem i ograniczaniem skutków ciężkiej choroby |
Załącznik nr 4
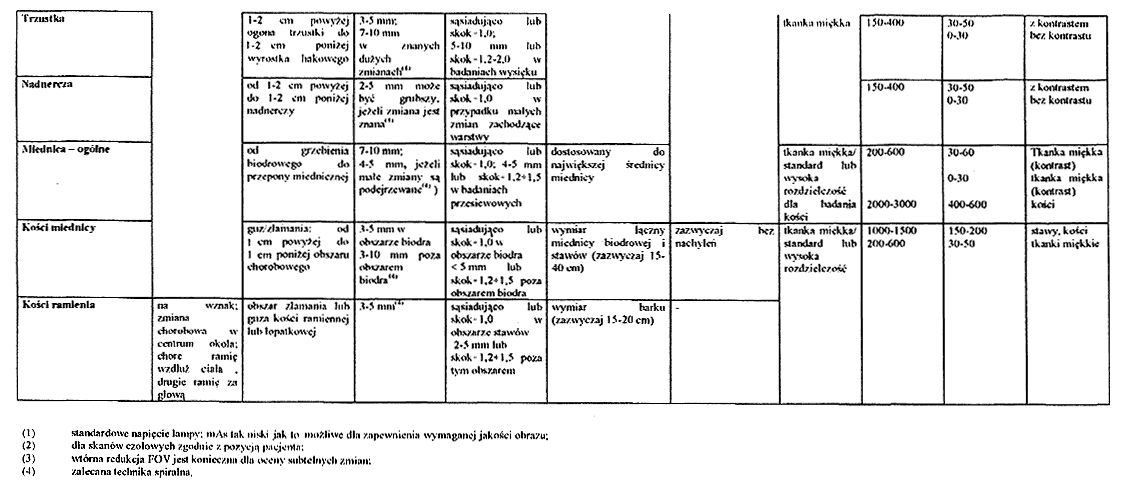
ZALECANE PODSTAWOWE PARAMETRY TECHNICZNE BADAŃ RENTGENODIAGNOSTYCZNYCH
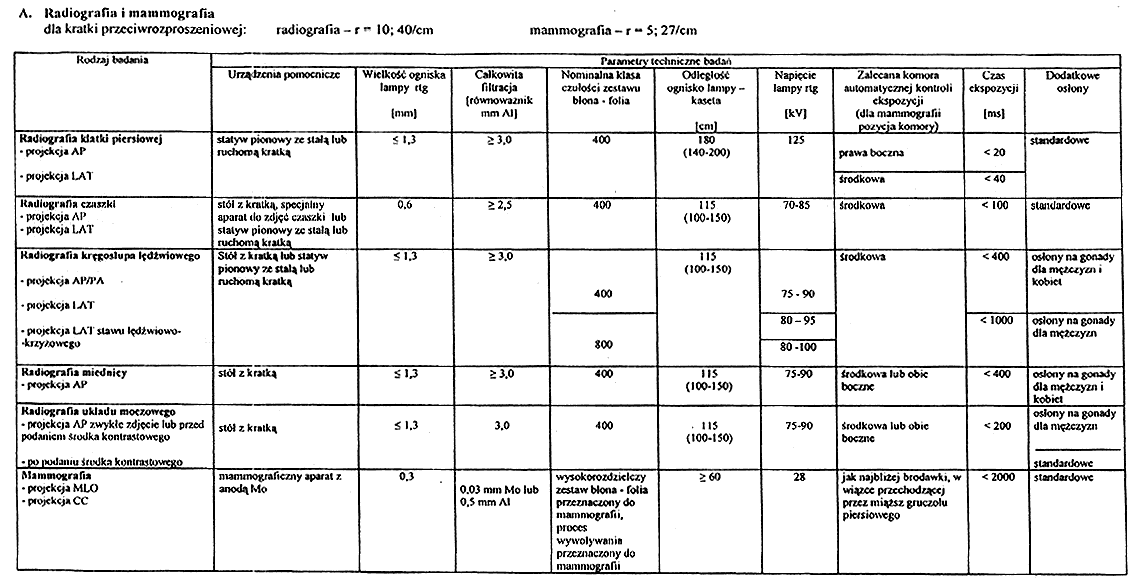
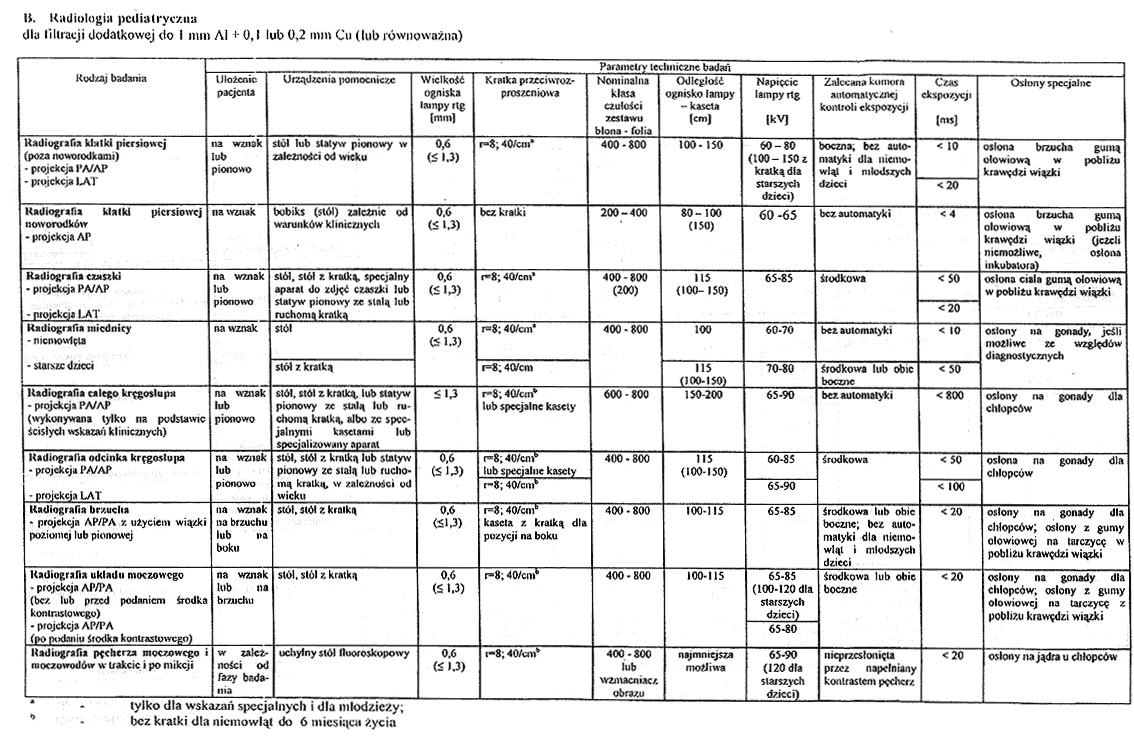
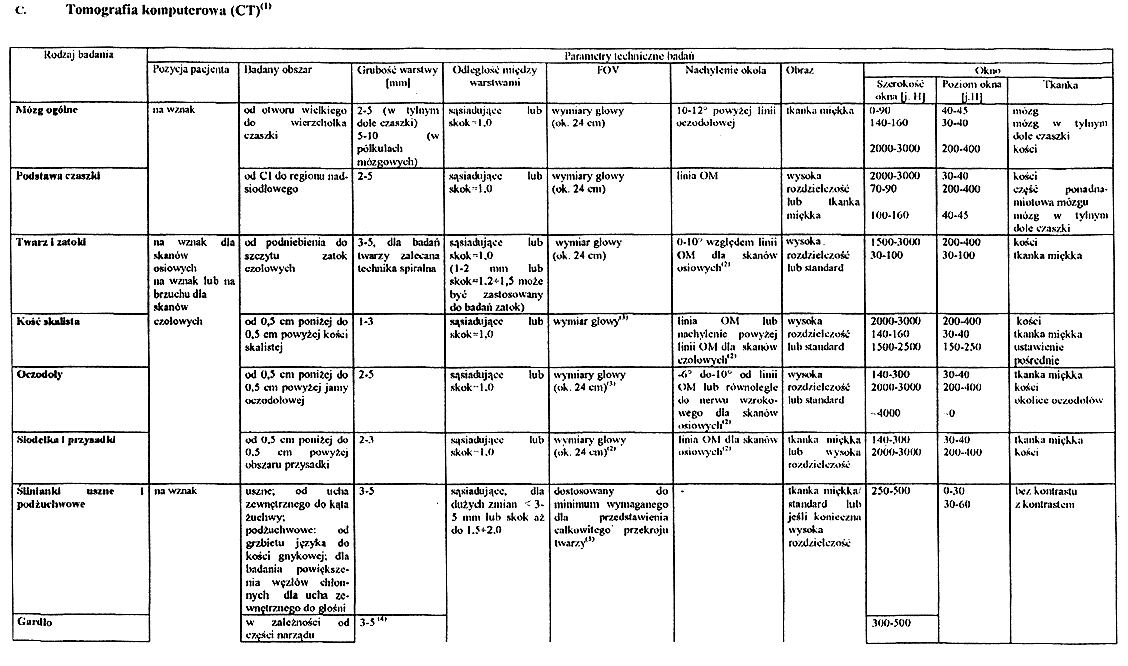
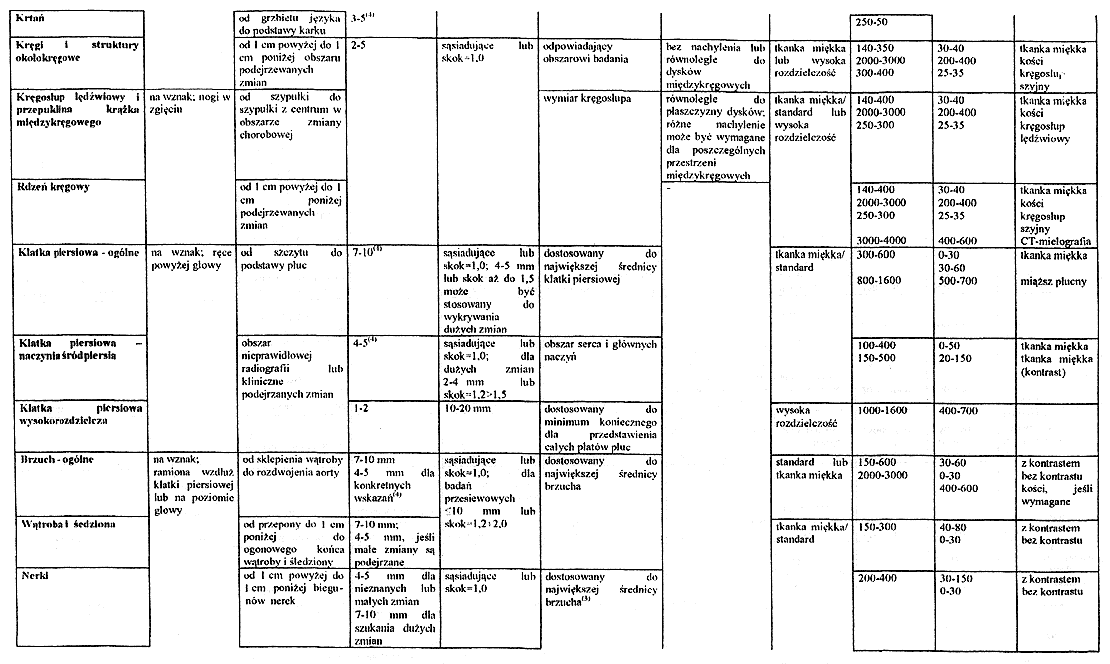
Załącznik nr 5
ZAKRES l CZĘSTOTLIWOŚĆ RUTYNOWYCH TESTÓW WEWNĘTRZNEJ KONTROLI PARAMETRÓW ORAZ ICH DOPUSZCZALNE ODCHYLENIA
|
| Rodzaj testu (pomiaru) | Częstotliwość wykonywania | Dopuszczalna tolerancja |
| 1 | Kontrola osiowości wiązki pierwotnej | raz na miesiąc (1/m-c) nie rzadziej niż 1/1000 zdjęć | <2% FFD |
| 2 | Kontrola temperatury i sensytometrycznych parametrów obróbki błon | codziennie | ±0.5°C |
| 3 | Kontrola przylegania błona-folia | 1/6 m-cy i po uszkodzeniach mechanicznych kasety | brak artefaktów |
| 4 | Badanie światłoszczelności ciemni i oświetlenia ochronnego | 1/3 m-ce i po uszkodzeniach mechanicznych |
|
| 5 | Badanie światłoszczelności kaset | 1 /m-c i po uszkodzeniach mechanicznych bądź stwierdzonych zadymieniach filmów |
|
| 6 | Ocena osłonności przepustów i pojemników na kasety | 1/12 m-cy i po dokonanych zmianach |
|
| 7 | Ocena negatoskopów | po każdorazowej wymianie źródła światła |
|
| 8 | Ocena oświetlenia górnego i zasłon okiennych | po dokonanych zmianach |
|
| 9 | Analiza zdjęć odrzuconych | po zgromadzeniu 50 zdjęć |
|
| 10 | Kontrola jakości obrazu fantomu referencyjnego dla mammografów | 1/tydzień | zgodnie z zaleceniami producenta |
| 11 | Kontrola stopnia wzmocnienia folii | 1/3 m-ce |
|
| 12 | Kontrola powtarzalności ekspozycji | 1/3 m-ce |
|
| 13 | Kontrola zgodności pola świetlnego z polem wiązki promieniowania | 1/m-c |
|
| 14 | Ocena kratki przeciwrozproszeniowej | 1/3 m-ce |
|
|
| Tomografia komputerowa testy 7, 9 oraz: |
|
|
| 15 | Pomiar jednorodności odwzorowania referencyjnego fantomu | 1/1 m-c | ± 4 J.H. |
| 16 | Ocena rozdzielczości wysokokontrastowej | 1/1 m-c |
|
| 17 | Ocena rozdzielczości niskokontrastowej i poziomu szumu | 1/1 m-c |
|
| 18 | Pomiar grubości warstwy | 1/1 m-c | ± 20 % |
| 19 | Ocena dokładności i powtarzalności ustawienia (ruchu) stała | 1/1 m-c | ± 0,2 cm |
| 20 | Ocena dokładności świetlnych wskaźników położenia | 1/1 m-c | ± 0,2 cm |
Załącznik nr 6
ZAKRES I CZĘSTOTLIWOŚĆ WYKONYWANIA ORAZ TOLERANCJA PARAMETRÓW TECHNICZNYCH APARATURY PODLEGAJĄCEJ ZEWNĘTRZNYM AUDYTOM KLINICZNYM
|
| Rodzaj testu | Częstość wykonywania | Tolerancja | |
|
| Radiografia podstawowa |
|
| |
| 1. | Ocena dokładności ustawienia napięcia | raz /rok i po wymianie lampy lub generatora | < 10% | |
| 2. | Ocena powtarzalności ustawienia napięcia | „ | „ | < 10% |
| 3. | Ocena dokładności czasu ekspozycji | raz/rok i po każdej naprawie elementów mogących mieć wpływ na ten parametr | < 10% | |
| 4. | Ocena liniowości narastania dawki w przeliczeniu na jednostkę natężenia prądu anodowego | raz/rok i po wymianie lampy lub generatora | < 10% | |
| 5. | Ocena zgodności dawki w przeliczeniu na jednostkę ładunku | „ | „ | < 10% |
| 6. | Kontrola poprawności działania systemu AEC | „ | „ | < 10% |
| 7. | Pomiar warstwy połówkowego osłabienia (HVL) | „ | „ | zgodnie z normą IEC |
| 8. | Ocena rozmiarów ognisk lampy | „ | „ | zgodnie z normą IEC |
| 9. | Ocena kontrastowości i ostrości obrazu przy użyciu specjalistycznych fantomów | „ | „ | zgodnie z normą IEC |
| 10. | Pomiar natężenia promieniowania przenikającego przez tubus lampy | raz/rok i po wymianie lampy lub generatora | w odległości 1 m | |
| 11. | Pomiar dawek otrzymywanych przez pacjentów dla 3 typowych badań rtg (dla radiografii) | raz/rok i po wymianie lampy lub generatora | poniżej wartości referencyjnych | |
|
| Fluoroskopia (testy w pozycjach 1-10 oraz dodatkowo) |
|
| |
| 12. | Ocena rozdzielczości wysokokontrastowej toru wizyjnego | raz/rok | zgodnie z normą IEC | |
| „ | zgodnie z normą IEC | |||
| 13. | Ocena rozdzielczości niskokontrastowej toru wizyjnego obrazowania oraz poziomu szumu | „ | poniżej wartości referencyjnych |
| 14. | Pomiar mocy dawki w wiązce pierwotnej padającej na pacjenta | „ | < 10% |
| 15. | Pomiar maksymalnej mocy dawki |
|
|
| 16. | Sprawdzenie automatycznego wyłącznika czasowego po upływie 5 min. ekspozycji | raz/rok |
|
| 17. | Ocena zniekształcenia obrazu |
|
|
|
| Mammografia (testy w pozycjach 1-10 oraz dodatkowo) | raz/rok | zgodnie z EUR 16263EN |
| 12. | Pomiary dawki powierzchniowej na fantomie akredytacyjnym | raz/rok | 130-200 N |
| 13. | Pomiar wartości i równomierności siły nacisku płyty dociskowej |
|
|
|
| Angiografia (testy w pozycjach 1-10 oraz dodatkowo) | raz/rok | zgodnie z normą IEC |
| 12. | Ocena rozdzielczości wysokokontrastowej toru wizyjnego | raz/rok | zgodnie z normą IEC |
| 13. | Ocena rozdzielczości niskokontrastowej toru wizyjnego oraz poziomu szumów | raz/rok |
|
| 14. | Pomiar maksymalnej mocy dawki w wiązce pierwotnej |
| poniżej wartości referencyjnych |
|
| Tomografia komputerowa (testy w pozycjach 1,2,3,7,8 oraz dodatkowo) |
|
|
| 9. | Pomiar jednorodności odwzorowania referencyjnego fantomu | raz/rok |
|
| 10. | Ocena poprawności odwzorowania kontrastu | raz/rok |
|
| 11. | Ocena rozdzielczości wysokokontrastowej | raz/rok | zgodnie z zaleceniami producenta |
|
|
| raz/rok |
|
| 12. | Ocena rozdzielczości niskokontrastowej i poziomu szumu | raz/rok | < 20% zg. z IEC 61223-2-6 |
|
|
|
| < 20% |
| 13. | Pomiar grubości warstwy | raz/rok |
|
|
|
|
| < poziomów |
| 14. | Ocena wielkości dozymetrycznych |
|
|
|
|
| raz/rok | referencyjnych 0.2 cm; ± 20 % od wyniku poprzed. |
| 15. | Ocena dokładności i powtarzalności ustawienia (ruchu) stołu |
| |
| raz/rok | ± 0,2 cm | ||
| 16. | Ocena dokładności świetlnych wskaźników położenia |
|
|
|
| Radiologia stomatologiczna (testy w pozycjach 1–10 oraz dodatkowo) |
|
|
| raz/rok |
| ||
|
|
|
| 20 cm dla U > 60 kV |
| 12. | Pomiar odległości FSD | raz/rok | 10 cm dla U < 60 kV |
|
| < 6cm | ||
| 13. | Pomiar średnicy pola na końcu tubusa |
|
|
| raz/rok |
| ||
| 14. | Ocena jednorodności obrazu |
| 3 |
Załącznik nr 7
OKRESY ZAPRZESTANIA KARMIENIA PIERSIĄ NIEMOWLĄT PO PODANIU PRODUKTÓW RADIOFARMACEUTYCZNYCH DLA CELÓW DIAGNOSTYCZNYCH
| Produkt radiofarmaceutyczny | Aktywność podana [MBq] | Okres po podaniu, w którym konieczne jest przerwanie karmienia [godz.] | ||
| 51Cr EDTA | 3 | 0 | ||
| 67 G- cytrynian | powyżej 150 | całkowite zaprzestanie karmienia | ||
| 99mTc – nadtechnecjan | 800 | 36 | ||
| „ | – | „ | 80 | 9 |
| „ | – mikrosfery | 80 | 9 | |
| „ | – erytrocyty, MIBI | 800 | 13 | |
| „ | – DTPA | 80 | 0 | |
| „ | – DMSA | 80 | 0 | |
| „ | – EC glukoheptonian | 800 | 0 | |
| „ | – fosfoniany | 600 | 0 | |
| 123I – jodki (czysty 123I)x | 20 | 5 | ||
| 123I – hipuran (czysty123I)x | 20 | 8 | ||
| 125I, 131I – wszystkie związki |
| całkowite zaprzestanie karmienia | ||
x– bez domieszki124I,125I
W przypadku stosowania produktów radiofarmaceutycznych, dla których nie jest konieczne zaprzestanie karmienia piersią, nie należy podawać dziecku pierwszej porcji pokarmu uzyskanej po podaniu związków promieniotwórczych.
Załącznik nr 8
LIMITY UŻYTKOWE DAWEK DLA PLANOWANIA OCHRONY PRZED PROMIENIOWANIEM OSÓB Z RODZINY PACJENTA LECZONEGO OTWARTYMI ŹRÓDŁAMI JODU-131 ORAZ OSÓB POSTRONNYCH
| Grupa osób | Limit użytkowy dawki |
| Dzieci do lat 10 oraz płody | 1 mSv |
| Dorośli do 60 roku życia | 3 mSv |
| Dorośli powyżej 60 roku życia | 15 mSv |
| Osoby postronne | 0,3 mSv |
Załącznik nr 9
MINIMALNY ZAKRES l CZĘSTOTLIWOŚĆ TESTÓW WEWNĘTRZNEJ KONTROLI JAKOŚCI W MEDYCYNIE NUKLEARNEJ
1. Mierniki aktywności bezwzględnej:
1) testy akceptacyjne sprawdzające zgodność parametrów i cech urządzenia z danymi katalogowymi. Ponadto sprawdzenie obecności atestu kalibracji dla poszczególnych radionuklidów;
2) testy eksploatacyjne:
a) codziennie – pomiar tła, pomiar odtwarzalności wskazań,
b) co miesiąc – testy na dokładność i precyzję pomiarów, testy na liniowość zależności pomiarów i aktywności,
c) co 2 lata – bezwzględna kalibracja urządzenia.
2. Kontrola jakości kamer scyntylacyjnych.
1) testy akceptacyjne.
Wszystkie kamery scyntylacyjne podlegają obowiązkowym testom akceptacyjnym na zgodność parametrów technicznych i cech ze specyfikacją producenta. Testy akceptacyjne obejmują również poprawność funkcjonowania komputerowych programów przetwarzania obrazów scyntygraficznych;
2) testy eksploatacyjne dla kamer planarnych:
a) codzienne:
– kalibracja energetyczna,
– pomiar tła (odtwarzalność),
– test na jednorodność czułości obszaru odwzorowywania detektora,
b) co miesiąc:
– kontrola jednorodności obrazowania przez detektor z zastosowaniem ilościowej analizy obrazu,
– kontrola przestrzennej liniowości i wewnętrznej rozdzielczości detektora (bar phantom test);
3) testy kontroli jakości rotacyjnych kamer scyntylacyjnych.
Kamery rotacyjne podlegają testom przewidzianym dla kamer planarnych, a ponadto następującym testom okresowym:
a) test jednorodności odwzorowywania przez detektor (duża liczba zarejestrowanych zliczeń) – częstotliwość zależna od stabilności detektora, przeciętnie raz w miesiącu,
b) test precyzji środka obrotu (z częstością zalecaną przez producenta),
c) test rozdzielczości tomograficznej (przy okazji testu precyzji środka obrotu),
d) test globalnego działania systemu (przy użyciu fantomu Jaszczaka – co kwartał),
e) test rozmiaru piksela matrycy obrazowej (co pół roku).
Wszystkie testy kontrolne obu typów kamer przeprowadza się po każdej naprawie urządzenia.
3. Kontrola jakości produktów radiofarmaceutycznych.
1) generatory nuklidów krótkożyjących. Testy kontrolne nie są obowiązkowe.
Należy sprawdzać wydajność elucji nuklidu.
Wskazane jest przeprowadzenie kontroli częstości radionuklidowej (zawartość 99Mo w eluacie z generatora molibdenowo-technetowego) i czystości chemicznej (zawartość AI+3) w przypadku trudności z uzyskaniem deklarowanej przez producenta wydajności elucji oraz w sytuacji, gdy uzyskiwane obrazy scyntygraficzne sugerują możliwość obecności w eluacie radionuklidu emitującego promieniowanie o energii fotonów wyższej niż fotonów emitowanych przez 994Tc;
2) produkty radiofarmaceutyczne:
a) gotowe do użycia nie podlegają kontroli jakości u użytkownika z wyjątkiem pomiaru aktywności podawanej pacjentom,
b) przygotowywane u użytkownika z zestawów do znakowania i eluatu z generatora.
Nie ma obowiązku rutynowej kontroli jakości wyznakowanego preparatu (z wyjątkiem pomiaru aktywności każdej porcji podawanej pacjentowi), jeżeli produkt radiofarmaceutyczny jest przygotowywany przez wykwalifikowanego pracownika i zgodnie z instrukcją producenta. Zalecane jest sprawdzanie czystości radiochemicznej wyznakowanego preparatu (ocena zawartości nadtechnecjanu i tlenku technetu zredukowanego, niewiązanego w formę kompleksu) w przypadku wątpliwości co do jakości przygotowywanego produktu radiofarma-ceutycznego,
c) pozostałe przygotowywane u użytkownika.
W przypadku znakowania komórek krwi bądź przygotowywania produktów radiofarmaceutycznych niezgodnie z instrukcją producenta, a także według własnych metod, obowiązują wszystkie wymagania dla producenta leków przewidziane przez prawo farmaceutyczne.
Załącznik nr 10
WYMAGANIA STAWIANE ZAKŁADOWI TELERADIOTERAPII W ZAKRESIE POMIESZCZEŃ I WYPOSAŻENIA
1. W każdym zakładzie teleradioterapii znajdują się:
1) pomieszczenia spełniające wymagania ochrony radiologicznej, w których będą zainstalowane megawoltowe aparaty terapeutyczne, symulator i ewentualnie tomograf komputerowy;
2) pomieszczenia towarzyszące wraz z infrastrukturą techniczną;
3) modelarnia;
4) pomieszczenia kliniczne;
5) pomieszczenia dla celów planowania leczenia.
2. Minimalne wyposażenie zakładu teleradioterapii stanowią:
1) co najmniej dwa megawoltowe aparaty terapeutyczne, w tym jeden akcelerator liniowy generujący wiązki elektronów o energii nie mniejszej niż 9 MeV. Integralną częścią każdego aparatu jest stół terapeutyczny i zestaw centratorów laserowych: jeden strzałkowy i dwa boczne;
2) co najmniej jeden symulator ze stołem terapeutycznym o identycznych parametrach technicznych jak stoły stosowane w leczeniu pacjenta i zestawem centratorów laserowych: jednym strzałkowym i dwoma bocznymi;
3) system planowania leczenia zintegrowany bezpośrednio lub pośrednio z tomografem komputerowym;
4) zestaw interfonii oraz interwizji, pozwalający na kontakt z pacjentem i jego obserwację podczas napromieniania;
5) specjalistyczna aparatura pomiarowo-kontrolna:
a) dwa dawkomierze z dwiema komorami jonizacyjnymi posiadającymi aktualne świadectwa legalizacji oraz jedno źródło kontrolne,
b) analizator pola promieniowania wiązek fotonów i elektronów,
c) fantomy wodne i fantomy stałe z adapterami dla posiadanych komór jonizujących,
d) atestowane przyrządy do pomiaru ciśnienia atmosferycznego i temperatury powietrza,
e) dawkomierze do codziennej kontroli dozymetrycznej, w ilości odpowiadającej liczbie akceleratorów,
f) zestaw akcesoriów i przyrządów do kontroli geometrii wiązek promieniowania w aparacie megawoltowym i symulatorze;
6) system wizualizacji wiązki zintegrowany z aparatem terapeutycznym lub co najmniej zestawy do wykonywania zdjęć sprawdzających, w ilości odpowiadającej liczbie megawoltowych aparatów terapeutycznych;
7) oprzyrządowanie do unieruchamiania i pozycjonowania pacjenta na stole terapeutycznym w pomieszczeniu symulatora i aparatów terapeutycznych.
3. Wyposażenie modelarni stanowią:
1) stół terapeutyczny i zestaw centratorów laserowych: strzałkowy i dwa boczne;
2) zestawy do wykonania masek unieruchamiających pacjenta, zapewniające odtwarzalność pozycji terapeutycznej pacjenta i centrowania wiązek promieniowania;
3) zestawy do przygotowania osłon narządów niebędących przedmiotem leczenia;
4) zestawy urządzeń do mocowania masek unieruchamiających dla każdego aparatu terapeutycznego.
4. Zakład teleradioterapii ma zagwarantowany, dla potrzeb planowania leczenia, dostęp do tomografu komputerowego.
Załącznik nr 11
WYMAGANIA DOTYCZĄCE WYPOSAŻENIA ZAKŁADU BRACHYTERAPII
1. Wyposażenie zakładu brachyterapii stanowią co najmniej:
1) jeden aparat do zdalnego wprowadzania źródeł promieniotwórczych z zestawem aplikatorów;
2) aparat rentgenowski do sprawdzania położenia aplikatorów, źródeł oraz wykonywania zdjęć lokalizacyjnych;
3) system do planowania leczenia w brachyterapii;
4) dwa dawkomierze z komorą jonizacyjną i fantom do pomiarów aktywności;
5) system do monitorowania dawki w czasie napromieniania mocą dawki większą od 12 Gy/h;
6) telefon, interfonia lub przycisk alarmowy umożliwiający kontakt pacjenta z punktem pielęgniarskim;
7) kamery telewizyjne oraz interfonia umożliwiające kontakt z pacjentem przy aparatach do napromieniania wysoką mocą dawki.
2. W przypadku aplikacji wymagających wykonania indywidualnych aplikatorów metodą odcisków lub odlewów zakład brachyterapii posiadać powinien możliwość korzystania z modelarni.
3. Aparaty terapeutyczne stosowane w zakładach brachyterapii, służące bezpośrednio do napromieniania pacjenta metodą zdalnego wprowadzania źródeł promieniotwórczych, muszą spełniać następujące wymagania:
1) wyłączenie i ponowne włączenie aparatu nie może likwidować sygnalizowanego błędu;
2) posiadanie systemu zabezpieczeń zapewniającego wycofanie źródeł promieniotwórczych do pojemnika ochronnego w przypadku awarii aparatu, awarii zasilania, przypadkowego przerwania leczenia lub wtargnięcia do pomieszczenia osoby nieleczonej.
4. Aparaty terapeutyczne do napromieniania wysoką mocą dawki stosowane w zakładach brachyterapii powinny ponadto:
1) posiadać dwa niezależne systemy odliczające czas i informujące o zakończeniu napromieniania;
2) realizować tylko leczenie zgodne z wybranym z konsoli sterującej;
3) weryfikować ustawione warunki i sygnalizować przypadkowe błędy personelu;
4) drukować w protokole leczenia wszystkie informacje o przeszkodach i błędach w realizacji leczenia;
5) być wyposażone w zestawy przyrządów do regularnego sprawdzania bezpiecznej pracy aparatu.
Załącznik nr 12
ZASADY, ZAKRES ORAZ CZĘSTOTLIWOŚĆ PRZEPROWADZANIA KONTROLI APARATÓW TERAPEUTYCZNYCH W ZAKŁADACH RADIOTERAPII
1. Warunkiem instalacji i dopuszczenia aparatu terapeutycznego do użytkowania po instalacji i w czasie eksploatacji jest dokonanie odbioru technicznego od producenta oraz okresowa kontrola parametrów mechanicznych i dozymetrycznych aparatu terapeutycznego w celu utrzymania ich wartości w dopuszczalnym zakresie.
2. Osobami odpowiedzialnymi za przeprowadzenie odbioru, o którym mowa w pkt 1, są pracownicy autoryzowanego serwisu aparaturowego oraz fizycy medyczni zatrudnieni w zakładzie radioterapii.
3. Osobami odpowiedzialnymi za przeprowadzenie okresowych kontroli są pracownicy serwisu, fizycy medyczni, specjaliści wspomagający fizyków medycznych i technicy radioterapii.
4. Pełna ocena parametrów technicznych akceleratora po instalacji obejmuje kontrolę:
1) wyłączników bezpieczeństwa;
2) awaryjnego licznika dawki;
3) położeń: 0°, 90°, 180° i 270° dla ruchu obrotowego ramienia i kolimatora akceleratora;
4) zgodność osi obrotu kolimatora z osią symulacji świetlnej;
5) izocentrum mechanicznego;
6) centratorów;
7) telemetru;
8) symulacji świetlnej pola promieniowania;
9) wyposażenia dodatkowego: kolimatorów wiązek elektronów, klinów mechanicznych, osłon i tac do osłon;
10) systemów interwencji i interfonii;
11) dodatkowych parametrów, które wynikają z indywidualnych uwarunkowań konstrukcyjnych danego aparatu.
5. Ocena parametrów dozymetrycznych akceleratora po instalacji obejmuje:
1) kontrolę pola promieniowania;
2) kontrolę jednorodności wiązki promieniowania;
3) kontrolę symetrii wiązki promieniowania;
4) określenie jakości wiązek promieniowania rentgenowskiego oraz energii wiązek elektronów;
5) wyznaczenie wydajności wiązki promieniowania;
6) kontrolę powtarzalności wydajności wiązki promieniowania;
7) kontrolę proporcjonalności dawki pochłoniętej do jednostek monitorowych;
8) kontrolę pracy akceleratora w różnych położeniach ramienia aparatu;
9) kontrolę stabilności wydajności wiązek promieniowania podczas dnia pracy;
10) kontrolę dodatkowych parametrów, które wynikają z indywidualnych uwarunkowań konstrukcyjnych danego przyspieszacza.
6. Okresowa kontrola akceleratorów obejmuje:
1) następujące testy codzienne wykonywane przez techników radioterapii:
a) kontrola systemu blokady drzwi wejściowych do akceleratora,
b) kontrola telemetru,
c) kontrola symulacji świetlnej pola promieniowania,
d) kontrola centratorów;
2) pomiary wydajności wiązek promieniowania dokonywane przez fizyków medycznych bądź specjalistów wspomagających fizyka medycznego co najmniej 2 razy w tygodniu;
3) cotygodniowe testy wykonywane przez pracowników serwisu aparatury;
a) kontrola telemetru,
b) kontrola symulacji świetlnej pola promieniowania,
c) kontrola centratorów,
d) wyposażenia dodatkowego: kolimatorów wiązek elektronów, klinów mechanicznych, osłon i tac do osłon;
4) następujące testy wykonywane raz na kwartał przez fizyków medycznych i pracowników serwisu aparatury;
a) kontrola świateł informacyjnych i ostrzegawczych,
b) kontrola wyłączników bezpieczeństwa,
c) awaryjnego licznika dawki,
d) położeń: 0°, 90°, 180° i 290° dla ruchów obrotowych ramienia i kolimatora akceleratora,
e) zgodności osi obrotu kolimatora z osią symulacji świetlnej,
f) izocentrum mechanicznego,
g) centratorów,
h) telemetru,
i) symulacji świetlnej pola promieniowania,
j) wyposażenia dodatkowego: kolimatorów wiązek elektronów, klinów mechanicznych, osłon i tac do osłon,
k) systemów interwizji i interfonii,
l) kontrola pola promieniowania;
5) następujące testy wykonywane co najmniej 2 razy w roku przez fizyków medycznych:
a) kontrola jednorodności wiązek promieniowania,
b) kontrola symetrii wiązek promieniowania,
c) kontrola jakości wiązek promieniowania rentgenowskiego oraz energii wiązek elektronów,
d) kontrola pracy akceleratorów w różnych położeniach ramienia aparatu;
6) następujące testy wykonywane co najmniej raz w roku przez fizyków medycznych bądź specjalistów ich wspomagających:
a) kontrola proporcjonalności dawki pochłoniętej do jednostek monitorowych,
b) kontrola stabilności wydajności wiązki promieniowania podczas dnia pracy.
7. W celu oceny parametrów technicznych aparatu kobaltowego po instalacji należy dokonać kontroli:
1) wyłączników bezpieczeństwa;
2) położenia 0° dla ruchów obrotowych ramienia, głowicy i kolimatorów aparatu;
3) zgodności osi obrotu kolimatora z osią świetlnej pola;
4) izocentrum;
5) centratorów;
6) telemetru;
7) symulacji świetlnej pola promieniowania;
8) wyposażenia dodatkowego: filtrów klinowych, osłon i tac do osłon;
9) zegara;
10) dodatkowych parametrów, które wynikają z indywidualnych uwarunkowań konstrukcyjnych danego aparatu.
8. W celu oceny parametrów dozymetrycznych aparatu kobaltowego po instalacji należy dokonać;
1) kontroli pola promieniowania;
2) pomiaru wydajności aparatu;
3) pomiaru „czasu martwego” przesuwu źródła.
9. Okresowa kontrola aparatów kobaltowych obejmuje:
1) następujące testy codzienne wykonywane przez techników radioterapii:
a) kontrola systemu blokady drzwi wejściowych do aparatu,
b) kontrola telemetru,
c) kontrola symulacji świetlnej pola promieniowania,
d) kontrola centratorów;
2) cotygodniowe testy wykonywane przez pracowników serwisu aparatury:
a) kontrola telemetru,
b) kontrola symulacji świetlnej pola promieniowania,
c) kontrola centratorów,
d) wyposażenia dodatkowego: filtrów klinowych, osłon i tac do osłon,
e) zegara;
3) testy wykonywane raz na kwartał przez fizyków medycznych i pracowników serwisu aparatury: wymienione w ust. 6;
4) testy wykonywane raz na kwartał przez fizyków medycznych i specjalistów wspomagających ich pracę: wymienione w ust. 7.
10. Po instalacji aparatu terapeutycznego fizycy medyczni dokonują pomiarów dozymetrycznych dla potrzeb systemu planowania leczenia wg protokołu zgodnie z wymaganiami danego systemu.
11. Okresowe przeglądy i konserwację akceleratorów przeprowadza autoryzowany serwis wg harmonogramu uzgodnionego pomiędzy użytkownikami i producentem akceleratora, co najmniej jeden raz na kwartał, a w przypadku bomby kobaltowej nie rzadziej niż jeden raz na pół roku.
12. Pracownicy serwisu i aparatury są upoważnieni do dokonywania przeglądów, konserwacji i codziennego uruchamiania akceleratora zgodnie z instrukcją obsługi aparatu i po odbyciu stosownych szkoleń.
Załącznik nr 13
WYMAGANIA PROCEDUR SYSTEMU ZARZĄDZANIA I KONTROLI JAKOŚCI RADIOTERAPII
1. System zarządzania i kontroli jakości w zakładzie radioterapii wdraża zespół powołany przez kierownika zakładu w składzie co najmniej dwóch lekarzy specjalistów radioterapii, dwóch fizyków medycznych i jeden starszy technik radioterapii.
2. System kontroli jakości radioterapii, zwany dalej „SKJR”, jest dokumentem zawierającym określenia celów i zadań zakładu radioterapii, oraz procedur i etapów leczenia promieniami, form i sposobów ich nadzoru oraz osób odpowiedzialnych za ich realizację w celu zapewnienia najwyższej precyzji i jakości leczenia promieniami indywidualnych pacjentów.
3. Procedura jest dokumentem opracowanym w formie instrukcji, zawierającym szczegółową informację i opis działań nią objętych oraz osób je realizujących, ze wskazaniem ich obowiązków, uprawnień, podległości organizacyjnej i merytorycznej oraz zakresu odpowiedzialności. Oprócz elementów technicznych i fizycznych procedura musi szczegółowo określić zasady postępowania z pacjentem, na etapie, którego procedura dotyczy.
4. Zbiór procedur w ramach SKJR, o których mowa w ust. 2 i 3, musi zawierać szczegółowe instrukcje dotyczące:
1) rejestracji pacjenta, założenia i prowadzenia dokumentacji lekarskiej i jej archiwizacji;
2) kryteriów kwalifikacji i zasad wyznaczenia terminów rozpoczęcia radioterapii i dopuszczalnego czasu oczekiwania na rozpoczęcie leczenia oraz zasad przestrzegania kolejności i ewentualnych priorytetów;
3) planowania objętości tarczowej oraz zasad ich weryfikacji i dopuszczalnych zakresów odchyleń;
4) wyboru i wykonania masek unieruchamiających oraz modyfikatorów rozkładu dawki promieniowania w wyznaczonych objętościach (filtry, kompensatory i inne);
5) symulacji i dwu- lub trójwymiarowego planowania radioterapii;
6) obowiązujących w zakładzie standardowych technik radykalnej i paliatywnej radioterapii oraz sposobów frakcjonowania dawki promieniowania dla różnych typów i lokalizacji nowotworów, jak również kryteria ich doboru dla indywidualnego pacjenta;
7) wypełnienia karty napromieniania, wyboru aparatu terapeutycznego oraz szczegółowy harmonogram realizacji radioterapii u indywidualnego pacjenta;
8) ułożenia i unieruchomienia pacjenta na stole terapeutycznym, centrowania wiązek promieniowania, codziennej kontroli, powtarzalności fizycznych i technicznych parametrów napromieniania w trakcie leczenia.
5. Każda z procedur, o których mowa w ust. 4, musi ponadto określić osobę lub osoby odpowiedzialne za ich realizację oraz zakres ich obowiązków.
6. System kontroli jakości radioterapii obejmuje również procedury w zakresie kalibracji i dozymetrii, serwisu i przeglądu aparatury terapeutycznej i wspomagającej, o których mowa w ust. 3–5.
7. SKJR musi uwzględnić i definiować wymagane postępowanie w przypadku nieprawidłowości lub błędów występujących przy realizacji procedur objętych systemem kontroli, a w szczególności:
1) sposoby i formę identyfikacji, zgłaszania i dokumentacji niezgodności (błędów) w realizacji procedury) objętych systemem kontroli;
2) sposoby zapobiegania i usuwania nieprawidłowości, o których mowa w pkt 1;
3) osoby i zakres ich odpowiedzialności w realizacji zadań, o których mowa w pkt 1 i 2.
8. SKJR musi uwzględniać i ściśle określać formę, zakres i częstość audytów (kontroli) procedur objętych systemem kontroli jakości, w tym co najmniej:
1) audyt kwalifikacji do leczenia promieniami;
2) audyt wyznaczania objętości zainteresowania;
3) audyt pozycjonowania i unieruchamiania pacjenta na stole terapeutycznym;
4) audyt symulacji radioterapii;
5) audyt planowania radioterapii (technika, sposób frakcjonowania);
6) audyt karty napromieniania;
7) audyt realizacji leczenia promieniami.
9. Audyt procedury polega na kontroli precyzji wykonania procedury lub etapach leczenia zgodnie z przyjętymi kryteriami i dopuszczalnym zakresie zdefiniowanym w dokumencie SKJR.
10. W zakładzie radioterapii obowiązują audyty wewnętrzne i zewnętrzne:
1) audyt wewnętrzny realizują u każdego pacjenta w oparciu o protokół audytów pracownicy wyznaczeni przez kierownika zakładu, którzy nie są bezpośrednio związani z jednym lub kolejnymi etapami planowania i realizacji radioterapii;
2) audyt zewnętrzny jest formą okresowej, całościowej lub wybiórczej kontroli zgodności wykonania procedur technicznych, fizycznych, terapeutycznych, dozymetrycznych z zapisem w dokumencie SKJR;
3) audyt zewnętrzny jest prowadzony zgodnie z odpowiednimi obowiązującymi przepisami lub na szczególne zalecenie ministra właściwego do spraw zdrowia oraz z inicjatywy kierownika zakładu w ramach jednego z programów europejskich.
11. System kontroli jakości radioterapii opracowuje indywidualnie każdy zakład radioterapii zgodnie z zakresem prowadzonej działalności w oparciu o zalecenia Raportu ICRU 60, 62 oraz raportu QART–ESTRO, 1995.
12. System kontroli jakości radioterapii jest otwartym dokumentem w zakładzie radioterapii i wymaga implementacji w przypadku zmian w strukturze organizacyjnej, wdrażania nowych technik, metod leczenia i zmian w wyposażeniu zakładu.
- Data ogłoszenia: 2002-12-31
- Data wejścia w życie: 2003-01-01
- Data obowiązywania: 2003-01-01
- Dokument traci ważność: 2005-10-21

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA