REKLAMA
Dziennik Ustaw - rok 2002 nr 196 poz. 1658
ROZPORZĄDZENIE MINISTRA ŚRODOWISKA1)
z dnia 6 listopada 2002 r.
w sprawie metodyk referencyjnych badania stopnia biodegradacji substancji powierzchniowoczynnych zawartych w produktach, których stosowanie może mieć wpływ na jakość wód.
Na podstawie art. 45 ust. 1 pkt 2 ustawy z dnia 18 lipca 2001 r. – Prawo wodne (Dz. U. Nr 115, poz.1229 i Nr 154, poz. 1803 oraz z 2002 r. Nr 113, poz. 984 i Nr 130, poz. 1112) zarządza się, co następuje:
1) metodykę wyodrębniania anionowych i niejonowych substancji powierzchniowoczynnych;
2) metodykę rozdziału anionowych i niejonowych substancji powierzchniowoczynnych;
3) metodykę prowadzenia procesu biodegradacji anionowych i niejonowych substancji powierzchniowoczynnych;
4) metodykę oznaczania stężenia anionowych substancji powierzchniowoczynnych;
5) metodykę oznaczania stężenia niejonowych substancji powierzchniowoczynnych;
6) metodykę obliczania stopnia biodegradacji anionowych i niejonowych substancji powierzchniowoczynnych.
2. Schemat wymaganej kolumny jonowymiennej do rozdziału anionowych i niejonowych substancji powierzchniowoczynnych zawiera załącznik nr 3 do rozporządzenia.
2. Schemat ideowy aparatury oraz podstawowych elementów konstrukcji aparatury wymaganej do prowadzenia procesu biodegradacji anionowych i niejonowych substancji powierzchniowoczynnych zawiera załącznik nr 5 do rozporządzenia.
2. Schemat aparatury wymaganej do zatężania i wypleniania niejonowych substancji powierzchniowoczynnych zawiera załącznik nr 8 do rozporządzenia.
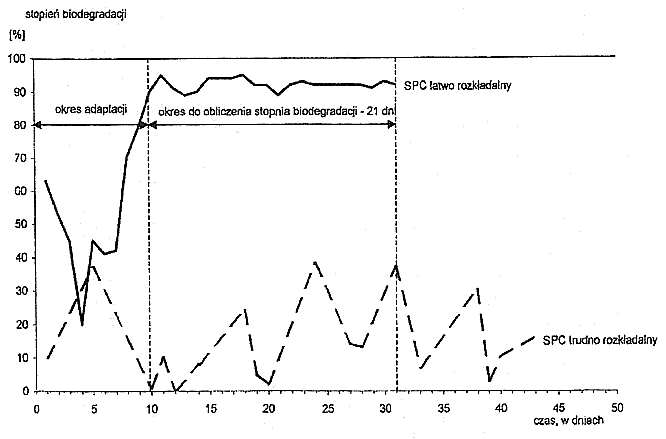
2. Wymagany prawidłowy przebieg procesu biodegradacji anionowych i niejonowych substancji powierzchniowoczynnych w czasie określa wykres, stanowiący załącznik nr 10 do rozporządzenia.
Minister Środowiska: S. Żelichowski
|
|
1) Minister Środowiska kieruje działem administracji rządowej – środowisko, na podstawie § 1 ust. 2 pkt 2 rozporządzenia Prezesa Rady Ministrów z dnia 20 czerwca 2002 r. w sprawie szczegółowego zakresu działania Ministra Środowiska (Dz. U. Nr 85, poz. 766).
Załączniki do rozporządzenia Ministra Środowiska
z dnia 6 listopada 2002 r. (poz. 1658)
Załącznik nr 1
METODYKA WYODRĘBNIANIA ANIONOWYCH l NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
1. Zasada metody
Metodę stosuje się do wyodrębniania anionowych i niejonowych substancji powierzchniowoczynnych. W celu wyodrębniania anionowych i niejonowych substancji powierzchniowoczynnych z jednorodnej próbki produktu należy wyekstrahować etanolem substancje powierzchniowoczynne. Otrzymany ekstrakt etanolowy należy odparować, po czym suchą pozostałość rozpuścić w wodnym roztworze 2-propanolu.
2. Wymagane odczynniki i aparatura
2.1. Odczynniki i roztwory
a. Woda dejonizowana lub destylowana.
b. Etanol, C2H5OH, 95% (v/v); (dopuszcza się stosowanie jako substancji skażającej metanolu lub 2-butanonu).
c. 2-propanol, CH3CHOHCH3, wodny roztwór 50% (v/v).
2.2. Wymagana aparatura i materiały
a. Kolba okrągłodenna o pojemności 2000 ml z korkiem szlifowanym i z chłodnicą zwrotną.
b. Aparat filtracyjny próżniowy o średnicy 90 mm do sączków papierowych, odporny na ogrzanie.
c. Kolba ssawkowa o pojemności 2000 ml.
d. Cylindry miarowe.
e. Zlewki.
f. Łaźnia wodna.
g. Suszarka próżniowa.
h. Wyparka obrotowa.
3. Wymagany sposób wykonania badania
Do kolby okrągłodennej należy wprowadzić 250 g wyrobu i dodać 1250 ml etanolu. Zamontować chłodnicę zwrotną i całość ogrzać do wrzenia, stosując łaźnię wodną, a następnie gotować pod chłodnicą zwrotną przez jedną godzinę. Następnie szybko przefiltrować etanolowy roztwór, stosując aparat filtracyjny próżniowy ogrzany uprzednio do temperatury 50°C, zbierając filtrat w kolbie ssawkowej. Przemyć kolbę i filtr próżniowy za pomocą około 200 ml gorącego etanolu, dołączając go do filtratu. Otrzymany filtrat etanolowy odparować do sucha, stosując do tego celu wyparkę obrotową lub inne powszechnie stosowane urządzenie do odparowywania. Operację ekstrakcji należy powtórzyć, jeśli potrzebna jest większa ilość ekstraktu. Suchą pozostałość należy rozpuścić w 5 000 ml wodnego roztworu 2-propanolu. Otrzymany roztwór służy do rozdziału anionowych i niejonowych substancji powierzchniowoczynnych. Roztwór powinien być przechowywany w temperaturze poniżej 5°C.
Uwaga:
Do badania biodegradacji potrzebne jest ok. 50 g anionowych substancji powierzchniowoczynnych lub ok. 25 g niejonowych substancji powierzchniowoczynnych. Należy wstępnie określić zawartość anionowych lub niejonowych substancji powierzchniowoczynnych w produkcie, według metodyk podanych w załącznikach nr 6 i 7 do rozporządzenia, aby określić ilość produktu, którą należy poddać operacji ekstrakcji. Ilość jednorazowo użytego produktu powinna być ograniczona do maksimum 2000 g.
Przy wyodrębnianiu anionowych substancji powierzchniowoczynnych, w przypadku produktów w postaci płynu lub pasty, należy sprawdzić, czy w 250-gramowej próbce znajduje się nie więcej niż 55 g anionowych substancji powierzchniowoczynnych i 35 g mydła. Próbki produktów w postaci płynu lub pasty należy przed ekstrakcją odparować do sucha. Otrzymaną suchą pozostałość należy rozpuścić w 2000 ml etanolu i postępować w sposób określony w pkt 3.
Przy wyodrębnianiu niejonowych substancji powierzchniowoczynnych, w przypadku produktów w postaci płynu lub pasty, należy sprawdzić, czy w 250-gramowej próbce znajduje się nie więcej niż 25 g anionowych substancji powierzchniowoczynnych i 35 g mydła. Próbki produktów w postaci płynu lub pasty należy przed ekstrakcją odparować do sucha. Otrzymaną suchą pozostałość należy rozpuścić w 500 ml etanolu i postępować w sposób określony w pkt 3.
W przypadku wyrobów sypkich o niskiej gęstości (mniejszej niż 300 g/l) należy zwiększyć proporcję etanolu w stosunku do próbki jak 20:1.
Załącznik nr 2
METODYKA ROZDZIAŁU ANIONOWYCH l NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
1. Zasada metody
Metodę stosuje się do rozdziału anionowych i niejonowych substancji powierzchniowoczynnych.
W celu rozdziału anionowych i niejonowych substancji powierzchniowoczynnych roztwór otrzymany w wyniku wyodrębnienia tych związków należy przepuścić przez układ składający się z silnie kwaśnego wymieniacza kationowego oraz makroporowatego wymieniacza anionowego. Układ ten należy ogrzać do temperatury 50°C (323 K), w celu zapobieżenia wytrącaniu się kwasów tłuszczowych.
Niejonowe substancje powierzchniowoczynne otrzymywane są bezpośrednio z odpływu przez odparowanie rozpuszczalnika (wodnego roztworu 2-propanolu).
Anionowe substancje powierzchniowoczynne wymywane są z kolumny anionowej jako sole amonowe, po uprzednim usunięciu kwasów tłuszczowych.
Kationowe substancje powierzchniowoczynne, które mogą zakłócić przebieg biodegradacji, oraz oznaczenia analityczne eliminowane są dzięki wychwyceniu przez jonowy wymieniacz kationowy, umieszczony w kolumnie znajdującej się powyżej kolumny z jonowym wymieniaczem anionowym.
2. Wymagane odczynniki i aparatura
2.1. Odczynniki i roztwory
a. Woda dejonizowana lub destylowana.
b. 2-propanol, CH3CHOHCH3, wodny roztwór 50% (v/v).
c. Dwutlenek węgla (CO2), etanolowy roztwór 0,1%, otrzymamy przez przepuszczenie dwutlenku węgla przez etanol przez okres 10 minut. Do tego celu należy stosować rurkę z wbudowaną kostką porowatą. Używać jedynie świeżo przygotowany roztwór dwutlenku węgla.
d. Wodorowęglan amonowy (NH4HC03) w postaci roztworu o stężeniu c = 0,3 mol/l w mieszaninie wody i 2-propanolu (60:40 v/v), otrzymany poprzez rozpuszczenie 0,3 mola wodorowęglanu amonowego w mieszaninie zawierającej 60 części objętościowych 2-propanolu i 40 części objętościowych wody, a następnie rozcieńczenie tej mieszaniny w kolbie miarowej do objętości 1000 ml.
e. Wymieniacz kationowy (KAT) – silnie kwaśna żywica kationowa o wielkości ziarna 50–100 mesh odporna na działanie alkoholu.
f. Wymieniacz anionowy (AAT) – makroporowata żywica anionowa o wielkości ziarna 70–150 mesh.
g. Kwas solny, roztwór 10% HCl (m/m).
2. 2. Aparatura i materiały
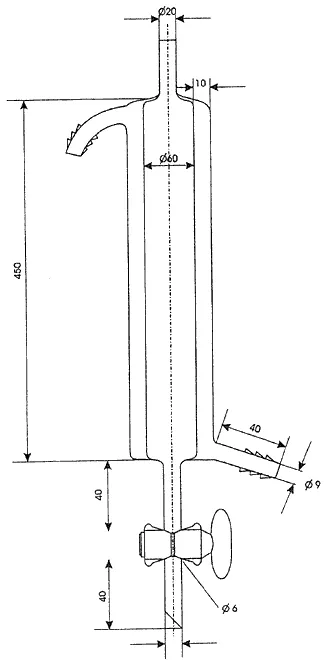
a. Kolumny jonowymienne z płaszczem grzejnym i kranem, o średnicy wewnętrznej 60 mm i wysokości 450 mm – 2 szt. (określone w załączniku nr 3 do rozporządzenia).
b. Cylindry miarowe.
c. Zlewki.
d. Łaźnia wodna.
e. Suszarka próżniowa.
f. Termostat.
g. Wyparka obrotowa.
3. Wymagany sposób wykonania badania
A. Przygotowanie kolumn jonowymiennych
Kolumna kationowa (KAT). W zlewce o pojemności 3000 ml należy umieścić 600 ml kationowej żywicy jonowymiennej, po czym dodać 2000 ml HCI i pozostawić na 2 godziny, mieszając od czasu do czasu. Następnie zdekantować kwas i przenieść żywicę do kolumny za pomocą wody zdejonizowanej. Kolumna powinna zawierać zatyczkę z waty szklanej. Należy przemywać kolumnę wodą dejonizowaną z szybkością 10–30 ml/min do momentu, gdy eluat będzie wolny od chlorków. Następnie należy zastąpić wodę wodnym roztworem 2-propanolu, przepuszczając go w ilości 2000 ml przez kolumnę z szybkością 10–30 ml/min. Po zakończeniu operacji kolumna jest przygotowana do pracy.
Kolumna anionowa (AAT). W zlewce o pojemności 3000 ml należy umieścić 600 ml anionowej żywicy jonowymiennej i dodać do niej 2000 ml wody dejonizowanej. Pozostawić żywicę do napęcznienia przez co najmniej 2 godziny, a następnie przenieść ją do kolumny za pomocą wody zdejonizowanej. Kolumna powinna zawierać zatyczkę z waty szklanej. Należy przemywać kolumnę 0,3 mol/l roztworem wodorowęglanu amonowego o objętości minimum 5000 ml do momentu, gdy eluat będzie wolny od chlorków. Następnie kolumnę przemyć za pomocą 2000 ml wody dejonizowanej, po czym zastąpić wodę wodnym roztworem 2-propanolu (2000 ml) i prowadzić przemywanie z szybkością 10–30 ml/min. Po zakończeniu operacji kolumna anionowa jest przeprowadzona w formę OH i jest przygotowana do pracy.
B. Procedura przeprowadzenia rozdziału
Należy połączyć kolumny jonowymienne tak, aby kolumna kationowa znajdowała się nad kolumną anionową. Ogrzać kolumny do temperatury 50°C, używając do jej kontroli termostatu. Następnie ogrzać 5000 ml roztworu otrzymanego – w sposób określony w załączniku nr 1 – w wyniku wyodrębniania substancji powierzchniowoczynnych do temperatury 60°C, i przepuścić go przez zestaw wymieniaczy jonowych z szybkością 20 ml/min. Kolumny przemyć za pomocą 1000 ml gorącego wodnego roztworu 2-propanolu.
W celu otrzymania wydzielonych niejonowych substancji powierzchniowoczynnych należy połączyć otrzymany eluat po przejściu przez zestaw kolumn oraz wodny roztwór 2-propanolu z przemywania, a następnie odparować do sucha na łaźni wodnej lub wyparce obrotowej. Pozostałość po odparowaniu zawiera niejonowe substancje powierzchniowoczynne. Do pozostałości dodać wody dejonizowanej do określonej objętości, odpowiedniej dla uzyskania stężenia odpowiedniego do oznaczeń ilościowych substancji niejonowych.
Otrzymany roztwór służy jako roztwór podstawowy do badania stopnia biodegradacji niejonowych substancji powierzchniowoczynnych. Roztwór powinien być przechowywany w temperaturze poniżej 5°C.
W celu otrzymania wydzielonych anionowych substancji powierzchniowoczynnych należy odłączyć od zestawu kolumnę kationową (KAT). Kolumnę anionową (AAT) należy przemyć 5000 ml roztworu dwutlenku węgla w etanolu, podgrzanego do temperatury 50°C, w celu wymycia kwasów tłuszczowych. Otrzymany eluat należy odrzucić. Następnie wymyć z kolumny AAT anionowe substancje powierzchniowoczynne za pomocą 5000 ml roztworu wodorowęglanu amonowego. Otrzymany w ten sposób eluat odparować do sucha na łaźni wodnej lub wyparce obrotowej. Pozostałość po odparowaniu zawiera anionowe substancje powierzchniowoczynne w postaci soli amonowych oraz ewentualnie inne substancje anionowe, niebędące substancjami powierzchniowoczynnymi, niemające wpływu na przebieg procesu biodegradacji. Do otrzymanej pozostałości należy dodać wody dejonizowanej w ilości koniecznej do uzyskania pożądanego stężenia anionowych substancji powierzchniowoczynnych.
Otrzymany roztwór służy jako roztwór podstawowy do badania stopnia biodegradacji niejonowych substancji powierzchniowoczynnych. Roztwór powinien być przechowywany w temperaturze poniżej 5°C.
C. Wymagany sposób regeneracji żywic jonowymiennych
Wymieniacz kationowy nie podlega regeneracji. Wymieniacz kationowy wyrzuca się po zakończeniu rozdziału.
Wymieniacz anionowy należy poddać regeneracji przez przepuszczenie 5000–6000 ml roztworu wodorowęglanu amonowego z szybkością 10 ml/min, aż do momentu, gdy eluat będzie wolny od substancji anionowych wykrywanych testem z błękitem metylenowym. Wymieniacz anionowy należy następnie przemyć za pomocą 2000 ml wodnego roztworu 2-propanolu przepuszczanego przez kolumnę z szybkością 10 ml/min. Kolumna anionowa jest przygotowana do ponownego użycia.
Załącznik nr 3
SCHEMAT WYMAGANEJ KOLUMNY JONOWYMIENNEJ DO ROZDZIAŁU ANIONOWYCH l NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
Załącznik nr 4
METODYKA PROWADZENIA PROCESU BIODEGRADACJI ANIONOWYCH l NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
1. Zasada metody
Metoda polega na prowadzeniu biodegradacji substancji powierzchniowoczynnych w postaci ich wodnego roztworu w laboratoryjnym zestawie, z wykorzystaniem osadu czynnego, symulującym pracę biologicznej oczyszczalni ścieków oraz codziennym oznaczaniu stężeń odpowiednio anionowych i niejonowych substancji powierzchniowoczynnych w dopływie, tj. w cieczy wprowadzanej do komory napowietrzania osadu czynnego, będącej ściekami syntetycznymi zawierającymi badane substancje powierzchniowoczynne oraz w odpływie, tj. w cieczy odprowadzanej z komory napowietrzania do odbieralnika po procesie osadzania osadu czynnego. Badanie należy prowadzić w cyklach 24-godzinnych przez czas obejmujący okres wstępnej adaptacji osadu czynnego oraz 21 dni po jego zakończeniu, przy czym okres wstępnej adaptacji osadu czynnego nie może przekraczać 6 tygodni.
Uwaga:
Pomieszczenie, w którym prowadzone są badania biodegradacji, powinno odpowiadać standardom laboratorium mikrobiologicznego. Podłoga i ściany powinny być wykonane z materiałów umożliwiających utrzymanie czystości. Ze względu na fakt, że osad czynny może zawierać organizmy patogenne, należy przedsięwziąć odpowiednie środki ostrożności w razie kontaktu z nim. Osoba obsługująca aparaty z osadem czynnym powinna nosić odpowiednie ubranie ochronne i rękawice.
Proces biodegradacji powinien być prowadzony w temperaturze od 19°C do 24°C w pomieszczeniu o ograniczonym dostępie światła i powietrzu wolnym od substancji mogących zakłócić przebieg badań.
2. Wymagane odczynniki i aparatura
2.1. Odczynniki i roztwory
a. Ścieki syntetyczne należy przygotować w następujący sposób:
W 1 000 ml wody wodociągowej rozpuścić:
– 160 mg peptonu,
– 110 mg ekstraktu mięsnego,
– 30 mg karbamidu (mocznika) – CO(NH2)2,
– 7 mg chlorku sodowego – NaCl,
– 4 mg uwodnionego chlorku wapniowego – CaCI2 x 2 H2O,
– 2 mg uwodnionego siarczanu magnezowego – MgSO4 x 7 H2O,
– 28 mg wodorofosforanu potasowego – K2HPO4,
– 20 mg +/– 2 mg anionowego związku powierzchniowoczynnego w przeliczeniu na dodecylobenzenosulfonian sodowy (oznaczanego jako MBAS) lub odpowiednio 10 mg +/– 1 mg niejonowego związku powierzchniowoczynnego w przeliczeniu na nonylofenol oksyetylenowany 10 molami tlenku etylenu (oznaczanego jako BiAS) – w zależności, jaki związek jest poddawany biodegradacji.
Substancje powierzchniowoczynne w postaci czystej mogą być stosowane w stanie oryginalnym. Substancje powierzchniowoczynne zawarte w produktach lub mieszaniny anionowych, kationowych lub niejonowych substancji powierzchniowoczynnych powinny zostać przed badaniem odpowiednio wyodrębnione i rozdzielone zgodnie z metodyką zawartą w załączniku nr 1 i 2 do rozporządzenia.
Ścieki syntetyczne powinny być przygotowywane codziennie.
b. Osad czynny świeżo pobrany z komory napowietrzania biologicznej oczyszczalni ścieków zasilanej głównie ściekami bytowymi.
W okresie między pobraniem osadu czynnego z oczyszczalni a jego użyciem osad powinien być przetrzymywany w warunkach tlenowych (napowietrzany).
c. Chlorek żelazowy, FeCI3, roztwór 5% (m/m).
2.2. Wymagana aparatura i materiały
a. Laboratoryjny zestaw do prowadzenia biodegradacji z wykorzystaniem osadu czynnego określony w załączniku nr 5 do rozporządzenia.
b. Lejki szklane o średnicy od 8 do 12 cm.
c. Suszarka laboratoryjna.
d. Sączki z bibuły filtracyjnej od 10 do 15 cm.
e. Filtr z włókna szklanego.
3. Wymagany sposób wykonania badania
3.1. Procedura wykonania badania
Komorę napowietrzania (C) i osadnik (D) należy napełnić ściekami syntetycznymi. Wysokość ustawienia osadnika (D) powinna być tak dobrana, aby komora (C) mieściła 3 l ścieków. Zaszczepienia dokonuje się przez wprowadzenie 3 ml osadu czynnego świeżo pobranego z komory napowietrzania biologicznej oczyszczalni ścieków zasilanej głównie ściekami bytowymi.
Do zbiornika (A) należy następnie wprowadzić 24 l ścieków syntetycznych. Przed rozpoczęciem procesu biodegradacji ze zbiornika (A) – tzw. dopływu pobrać około 200 ml próbki w celu oznaczenia stężenia anionowych lub niejonowych substancji powierzchniowoczynnych. Następnie uruchomić aparat do napowietrzania (G), powietrzny podnośnik cieczy (E) i pompę dozującą (B). Szybkość dopływu ścieków syntetycznych ze zbiornika (A) do komory napowietrzania (C) należy utrzymywać na poziomie 1 l/godz., co zapewni trzygodzinny czas napowietrzania i kontaktu badanej substancji z osadem czynnym.
Szybkość dopływu powietrza do komory napowietrzania (C) należy ustawić na takim poziomie, aby zapobiec osadzaniu się napowietrzonego osadu czynnego i aby zawartość rozpuszczonego tlenu wynosiła co najmniej 2 mg/l. Powietrzny podnośnik cieczy (E) powinien być tak ustawiony, aby osad czynny z osadnika (D) stale i regularnie krążył między osadnikiem (D) a komorą napowietrzania (C). Osad czynny zbierający się na brzegach komory napowietrzania (C), na dnie osadnika (D) i w przewodach należy zawracać co najmniej raz dziennie do obiegu przy pomocy szczotki. W przypadku gdy osad czynny nie osadza się w osadniku, można zwiększyć jego zdolność do osadzania (gęstość) przez dodanie 2 ml 5% roztworu chlorku żelazowego, powtarzając ten zabieg w razie potrzeby.
Ścieki z osadnika (D) należy gromadzić w odbieralniku (F) w ciągu 24 godzin, po czym po dokładnym wymieszaniu zawartości zbiornika pobrać około 200 ml próbki tzw. odpływu w celu oznaczenia stężenia pozostałych nierozłożonych anionowych lub niejonowych substancji powierzchniowoczynnych. Próbki należy analizować bezpośrednio po pobraniu zgodnie z metodyką określoną w załącznikach nr 6 i 7 do rozporządzenia, wykonując oznaczenia z dokładnością do 0,1 mg/l. Jeśli nie jest możliwe wykonanie badania bezpośrednio po pobraniu próbki, to próbkę należy zakonserwować, najlepiej przez zamrożenie.
Zbiornik (A) i odbieralnik (F) należy dokładnie umyć, po czym do zbiornika (A) wprowadzić ponownie 24 l ścieków syntetycznych i powtórzyć opisany powyżej proces biodegradacji.
Badanie należy prowadzić do momentu, gdy po wstępnej adaptacji osadu czynnego okres, w którym otrzymuje się wyrównane wyniki stopnia biodegradacji, wynosi 21 dni.
3.2. Kontrola przebiegu procesu biodegradacji
Codzienna kontrola przebiegu procesu biodegradacji powinna obejmować sprawdzanie intensywności dopływu ścieków surowych, stężenia tlenu w komorze napowietrzania oraz badania fizyczno-chemiczne próbek jednorazowych ścieków surowych i średniodobowych próbek ścieków oczyszczonych.
Codzienna kontrola fizyczno-chemiczna ścieków powinna obejmować pH oraz stężenie substancji powierzchniowoczynnych.
Okresowa kontrola, co najmniej dwa razy w tygodniu, obejmuje:
– oznaczanie w ściekach surowych i oczyszczonych: ChZT oznaczane wg normy PN-74/C-04578/03 lub PN-EN ISO 8467:2001 albo rozpuszczalny węgiel organiczny (RWO) oznaczany wg normy PN-ISO 9439+AC1,
– oznaczanie w osadzie czynnym: stężenia zawiesiny wg PN-EN 872 i indeksu objętościowego wg PN-75/C-04616/03.
Oznaczanie stężenia tlenu w komorze napowietrzania wykonuje się wg PN-EN 25814.
Sprawdzianem efektywności procesu biodegradacji jest wielkość chemicznego zapotrzebowania tlenu (ChZT) albo wielkość rozpuszczalnego węgla organicznego (RWO) w przesączach, uzyskiwanych po filtracji przez filtr z włókna szklanego próbek pobranych ze zbiornika (A) i odbieralnika (F). Spadek wartości ChZT lub RWO powinien ulegać zahamowaniu w momencie, kiedy oznaczenia stężenia anionowych lub niejonowych substancji powierzchniowoczynnych osiągną w przybliżeniu wartość stałą. Zakończenie okresu wstępnej adaptacji osadu czynnego poznaje się po stałości przebiegu krzywej pomiarów spadku ChZT lub RWO i pomiarów dziennej wartości stopnia biodegradacji, zgodnie z wykresem określonym w załączniku nr 10 do rozporządzenia.
Stężenie zawiesiny osadu czynnego w komorze (C) powinno wynosić ok. 2,5 g/l. W przypadku gdy jest ona wyższa, w celu zapobieżenia zakłóceniu równowagi biologicznej, należy usunąć partiami z układu nadmiar osadu czynnego.
Załącznik nr 5
SCHEMAT IDEOWY APARATURY ORAZ PODSTAWOWYCH ELEMENTÓW KONSTRUKCJI APARATURY WYMAGANEJ DO PROWADZENIA PROCESU BIODEGRADACJI ANIONOWYCH l NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
Rys. 1. Schemat ideowy aparatury wymaganej do prowadzenia procesu biodegradacji anionowych i niejonowych substancji powierzchniowoczynnych
Oznaczenia:
A. zbiornik na ścieki syntetyczne
B. pompa dozująca
C. komora napowietrzania
D. osadnik
E. powietrzny podnośnik cieczy
F. odbieralnik
G. aparat do napowietrzania (kostka napowietrzająca)
H. regulator dopływu powietrza
Zbiornik (A) i odbieralnik (F) powinny być wykonane ze szkła lub odpowiedniego tworzywa.
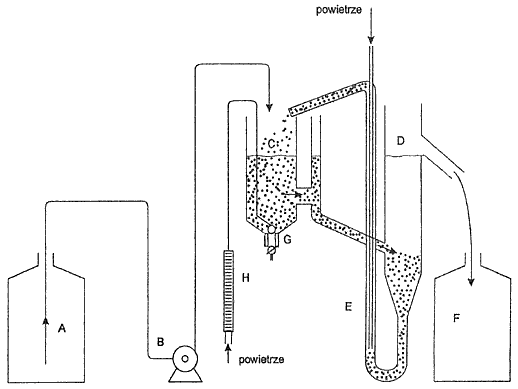
Rys. 2. Schemat podstawowych elementów konstrukcji aparatury wymaganej do prowadzenia procesu biodegradacji anionowych i niejonowych związków powierzchniowoczynnych
Załącznik nr 6
METODYKA OZNACZANIA STĘŻENIA ANIONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
Metoda l
1. Zasada metody
Metoda oznaczania stężenia anionowych substancji powierzchniowoczynnych polega na przeprowadzeniu ich reakcji z kationowym barwnikiem – tj. błękitem metylenowym w celu otrzymania barwnego kompleksu o niebieskim zabarwieniu, który wyekstrahowuje się z roztworu za pomocą chloroformu. Pomiar absorbancji ekstraktu chloroformowego przy długości fali 650 nm jest podstawą do określenia stężenia anionowych substancji powierzchniowoczynnych. Stężenie anionowych substancji powierzchniowoczynnych podaje się w przeliczeniu na dodecylobenzenosulfonian sodowy (MBAS).
2. Wymagane odczynniki i aparatura
2.1. Odczynniki i roztwory
a. Roztwór buforowy o pH 10, otrzymany poprzez rozpuszczenie 24 g wodorowęglanu sodowego (NaHCO3), czystego do analiz, i 27 g bezwodnego węglanu sodowego (Na2CO3), czystego do analiz, w wodzie dejonizowanej, a następnie rozcieńczenie wodą w kolbie miarowej do objętości 1 000 ml.
b. Błękit metylenowy w postaci roztworu obojętnego, otrzymanego poprzez rozpuszczenie 0,35 g błękitu metylenowego czystego do analiz w wodzie dejonizowanej i rozcieńczenie wodą dejonizowaną w kolbie miarowej do objętości 1000 ml. Roztwór należy przygotować co najmniej 24 godziny przed użyciem. Wartość absorbancji ślepej próby fazy chloroformowej mierzona w stosunku do użytego chloroformu nie może przekraczać przy długości fali 650 nm 0,015 dla warstwy pomiarowej o grubości 1 cm.
c. Błękit metylenowy, w postaci roztworu kwaśnego, otrzymanego poprzez rozpuszczenie 0,35 g błękitu metylenowego czystego do analiz w 500 ml wody dejonizowanej i zmieszanie z 6,5 mi kwasu siarkowego (H2SO4) o gęstości 1,84 g/ml, a następnie rozcieńczenie wodą dejonizowaną w kolbie miarowej do objętości 1000 ml. Roztwór należy przygotować co najmniej 24 godziny przed użyciem. Wartość absorbancji ślepej próby fazy chloroformowej mierzona w stosunku do użytego chloroformu nie może przekraczać przy długości fali 650 nm 0,015 dla warstwy pomiarowej o grubości 1 cm.
d. Chloroform (trójchlorometan) świeżo destylowany.
e. Dodecylobenzenosulfonian metylu, (C12H25C6H4SO3CH3).
f. Wodorotlenek potasowy, etanolowy roztwór o stężeniu KOH c = 0,1 mol/l.
g. Etanol, (C2H5OH), czysty.
h. Kwas siarkowy (H2SO4), wodny roztwór o stężeniu 0.5 mol/l.
i. Fenoloftaleina w postaci roztworu etanolowo-wodnego, otrzymanego poprzez rozpuszczenie 1 g fenoloftaleiny w 50 ml etanolu i w 50 ml wody dejonizowanej. Otrzymany roztwór należy odsączyć od osadu.
j. Kwas solny, (HCI), czysty do analiz, w postaci roztworu metanolowego otrzymanego przez zmieszanie 250 ml kwasu solnego i 750 ml metanolu.
2.2. Wymagana aparatura i materiały
a. Rozdzielacz o pojemności 250 ml.
b. Kolba miarowa o pojemności 50 ml.
c. Kolba miarowa o pojemności 500 ml.
d. Kolba miarowa o pojemności 1000 ml.
e. Kolba okrągłodenna o pojemności 250 ml, z zamknięciem szlifowanym i chłodnicą zwrotną wraz z kamykami wrzennymi.
f. Pehametr.
g. Fotometr przystosowany do pomiaru przy długości fali 650 nm, wyposażony w naczynka pomiarowe o grubości od 1 do 5 cm.
h. Bibuła filtracyjna jakościowa.
3. Wymagany sposób wykonania badania
3.1. Pobieranie próbek
Próbki nie powinny być pobierane przez warstwę piany. Próbki pobrane ze zbiornika (A) i odbieralnika (F) powinny być przefiltrowane i poddane badaniom natychmiast po pobraniu. W trakcie filtrowania należy odrzucić pierwsze 100 ml filtratu.
Uwaga:
Po umyciu urządzeń wodą należy je starannie wypłukać metanolowym roztworem kwasu solnego i ponownie wodą.
3.2. Wymagany sposób wykonania oznaczenia
Należy umieścić odmierzoną objętość próbki, jeśli zachodzi potrzeba – zobojętnionej, w rozdzielaczu o pojemności 250 mi. Objętość próbki powinna być tak dobrana, aby zawierała od 20 do 150 mg anionowych substancji powierzchniowoczynnych w przeliczeniu na dodecylobenzenosulfonian sodowy (MBAS). W przypadku niskich stężeń MBAS można stosować do 100 ml próbki. W przypadku gdy objętość próbki jest mniejsza niż 100 ml, należy ją rozcieńczyć wodą dejonizowaną do tej objętości.
Do 100 ml próbki dodać 10 ml roztworu buforowego, 5 ml obojętnego roztworu błękitu metylenowego i 15 ml chloroformu (roztwór alkaliczny). Wstrząsać zawartością rozdzielacza niezbyt energicznie przez 1 minutę w celu jej ujednorodnienia. Po rozdzieleniu się faz przenieść warstwę chloroformową do drugiego rozdzielacza zawierającego 110 ml wody dejonizowanej i 5 ml kwaśnego roztworu błękitu metylenowego (roztwór kwaśny). Wstrząsać mieszaninę przez 1 minutę i przefiltrować warstwę chloroformową do kolby pomiarowej przez sączek z waty uprzednio umyty i nasączony chloroformem.
Następnie należy wykonać jeszcze dwukrotnie ekstrakcję obu roztworów (alkalicznego i kwaśnego), stosując po 10 ml chloroformu. Przesączyć połączone ekstrakty chloroformowe przez zastosowany poprzednio sączek z waty i rozcieńczyć chloroformem z przemycia sączka w kolbie miarowej do objętości 50 ml. Wykonać pomiar absorbancji barwnego roztworu chloroformowego przy długości fali 650 nm, dobierając odpowiednią grubość naczyńka pomiarowego w zakresie od 1 do 5 cm. Pomiar wykonuje się wobec chloroformu jako odnośnika. Równolegle należy wykonać ślepą próbę.
3.3. Wymagany sposób sporządzenia krzywej wzorcowej
Roztwór wzorcowy należy przygotować, wykorzystując dodecylobenzenosulfonian metylu po jego zmydleniu do soli potasowej. Stężenie anionowych substancji powierzchniowoczynnych określa się w przeliczeniu na dodecylobenzenosulfonian sodowy o masie cząsteczkowej 348 (MBAS).
W kolbie okrągłodennej należy odważyć za pomocą pipety wagowej z dokładnością do 0,1 mg od 400 do 450 mg dodecylobenzenosulfonianu metylu. Dodać 50 ml etanolowego roztworu wodorotlenku potasowego i kilka kamyków wrzennych. Po zamontowaniu chłodnicy zwrotnej gotować zawartość kolby przez godzinę. Po oziębieniu przemyć chłodnicę i szlif za pomocą 30 ml etanolu, a popłuczyny dodać do kolby okrągłodennej. Roztwór miareczkować za pomocą kwasu siarkowego wobec fenoloftaleiny aż do zaniku barwy.
Otrzymany roztwór przenieść do kolby miarowej i rozcieńczyć wodą dejonizowaną do objętości 1000 ml.
Pobrać 25 ml roztworu i mieszając rozcieńczyć wodą dejonizowaną w kolbie miarowej do objętości 500 ml.
Stężenie anionowych substancji powierzchniowoczynnych w przeliczeniu na dodecylobenzenosulfonian sodowy (tzw. MBAS) w tak przygotowanym roztworze wzorcowym cMBASwz, w mg MBAS/ml, oblicza się wg wzoru:
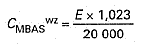
w którym:
E – oznacza masę próbki w mg.
W celu przygotowania krzywej wzorcowej należy pobrać: 1, 2, 4, 6 i 8 ml roztworu wzorcowego, rozcieńczyć wodą dejonizowaną w kolbach miarowych do objętości 100 ml, co odpowiada stężeniu MBAS odpowiednio: 0,01xcMBASwz , 0,02xcMBASwz, 0,04xcMBASwz, 0,06xcMBASwz, 0,08xcMBASwz. Następnie postępować ze 100 ml próbkami zgodnie z procedurą określoną w pkt 3.2, łącznie z etapem wykonania ślepej próby.
Następnie należy sporządzić krzywą wzorcową lub obliczyć współczynniki równania regresji, określające zależność absorbancji od stężenia anionowej substancji powierzchniowoczynnej w mg MBAS/ml.
4. Wymagany sposób obliczania wyników.
Stężenie anionowych substancji powierzchniowoczynnych w przeliczeniu na dodecylobenzenosulfonian sodowy (MBAS) w próbce analitycznej odczytuje się z krzywej wzorcowej lub oblicza z równania regresji.
Stężenie anionowej substancji powierzchniowoczynnej XMBAS, w mg/l, oblicza się wg wzoru:
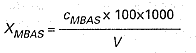
w którym:
cMBAS – oznacza stężenie anionowych substancji powierzchniowoczynnych w przeliczeniu na dodecylobenzenosulfonian sodowy odczytane z krzywej wzorcowej, w mg/ml,
V – oznacza objętość użytej próbki w ml.
Wyniki należy podawać z dokładnością do 0,1 mg/l.
W zależności od miejsca pobrania próbki XMBAS stanowi odpowiednio D lub O przy obliczaniu dziennej wartości stopnia biodegradacji,
gdzie:
D – oznacza stężenie anionowych lub niejonowych substancji powierzchniowoczynnych w tzw. dopływie (oznaczanych odpowiednio jako MBAS lub BiAS) w mg/l,
O – oznacza stężenie anionowych lub niejonowych substancji powierzchniowoczynnych w tzw. odpływie (oznaczanych odpowiednio jako MBAS lub BiAS) w mg/l.
Metoda II
Stężenie anionowych substancji powierzchniowoczynnych oznacza się metodą przepływową wg PN-C-04645, załącznik B.
Załącznik nr 7
METODYKA OZNACZANIA STĘŻENIA NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
1. Zasada metody
Metoda polega na zatężaniu i wypienieniu niejonowych substancji powierzchniowoczynnych z fazy wodnej do rozpuszczalnika organicznego działaniem strumienia gazu, usunięciu rozpuszczalnika organicznego i przeprowadzeniu niejonowych substancji powierzchniowoczynnych w trudno rozpuszczalny związek kompleksowy za pomocą zmodyfikowanego odczynnika Dragendorffa – tj. mieszaniny jodobizmutynu potasowego, chlorku barowego i lodowatego kwasu octowego (KbiJ4 + BaCI2 + CH3COOH). Osad odsącza się, przemywa kwasem octowym lodowatym i rozpuszcza w roztworze winianu amonowego. Obecne w roztworze jony bizmutu odmiareczkowuje się potencjometrycznie roztworem pirolidynoditiokarbaminianu sodowego przy pH 4–5 z zastosowaniem elektrody platynowej jako elektrody wskaźnikowej i kalomelowej lub chlorosrebrowej jako elektrody odniesienia. Stężenie niejonowych substancji powierzchniowoczynnych podaje się w przeliczeniu na nonylofenol oksyetylenowany 10 molami tlenku etylenu (BiAS).
Uwaga:
1. Metodę stosuje się do oznaczania niejonowych substancji powierzchniowoczynnych posiadających w cząsteczce od 6 do 30 grup tlenku etylenu.
2. Zawartość niejonowych substancji powierzchniowoczynnych w analizowanej próbce powinna wynosić od 250 do 800 mg.
2. Wymagane odczynniki i aparatura
2.1. Odczynniki
Uwaga:
Do sporządzania roztworów wszystkich odczynników stosuje się wodę dejonizowaną.
a. Octan etylu, (CH3COOC2H5), czysty do analiz, świeżo destylowany.
b. Wodorowęglan sodowy, (NaHCO3), czysty do analiz.
c. Kwas solny, (HCl), w postaci roztworu rozcieńczonego, otrzymanego przez rozcieńczenie 20 ml stężonego kwasu solnego wodą dejonizowaną w kolbie miarowej do objętości 1000 ml.
d. Metanol, (CH3OH), czysty do analiz, świeżo destylowany, przechowywany w butli szklanej.
e. Purpura bromokrezolowa w postaci roztworu, otrzymanego przez rozpuszczenie 0,1 g barwnika w 100 ml metanolu.
f. Kwas octowy lodowaty, (CH3COOH), o stężeniu 99–100%.
g. Zmodyfikowany odczynnik Dragendorffa, będący mieszaniną 2 objętości roztworu A i 1 objętości roztworu B. Mieszaninę tę należy przechowywać w butelce z brunatnego szkła, nie dłużej niż 1 tydzień.
– Roztwór A: otrzymany przez rozpuszczenie 1,7 g zasadowego azotanu bizmutu (III), BiON03 x H2O, czystego do analiz, w 20 ml kwasu octowego lodowatego i rozcieńczenie wodą do 100 ml. Następnie należy rozpuścić 65 g jodku potasowego, (KJ), czystego do analiz, w 200 ml wody. Następnie należy zmieszać oba roztwory w kolbie miarowej, dodać 200 ml kwasu octowego lodowatego i rozcieńczyć wodą do objętości 1000 ml.
– Roztwór B: otrzymany przez rozpuszczenie 290 g dwuwodnego chlorku baru (BaCI2 x 2 H2O) w wodzie i rozcieńczyć wodą w kolbie miarowej do objętości 1000 ml.
h. Winian amonowy w postaci roztworu otrzymanego przez połączenie 12,4 g kwasu winowego ((CHOHCOOH)2), czystego do analiz, i 12,4 ml ok. 25% roztworu amoniaku (NH3), czystego do analiz, a następnie rozcieńczenie wodą w kolbie miarowej do objętości 1000 ml. Można również użyć odpowiednią ilość winianu amonowego ((CHOHCOONH4)2), czystego do analiz.
i. Amoniak w postaci roztworu, otrzymanego przez rozcieńczenie wodą 40 ml roztworu amoniaku o gęstości 0,91 g/ml w kolbie miarowej do objętości 1000 ml.
j. Standardowy bufor octanowy, otrzymany przez rozpuszczenie 40 g wodorotlenku sodowego (NaOH), czystego do analiz, w 500 ml wody w zlewce i oziębienie, a następnie dodanie 120 ml kwasu octowego lodowatego i rozcieńczenie wodą do objętości1000 ml.
k. Pirolidynoditiokarbaminian sodowy (C5H8NNaS2) w postaci roztworu o stężeniu 0,0005 mol/l, otrzymanego przez rozpuszczenie 103,0 mg soli sodowej kwasu pirolidynoditiokarbaminowego (C5H8NNaS2 x 2 H2O) w 500 ml wody i dodaniu 10 ml 1-pentanolu, (C5H11OH), czystego do analiz, 0,5 g wodorowęglanu sodowego (NaHCO3), czystego do analiz, i rozcieńczenie wodą w kolbie miarowej do objętości 1 000 ml.
l. Siarczan miedziowy w postaci wzorcowych roztworów:
– Roztwór podstawowy o stężeniu 0,005 mol/l, otrzymany przez zmieszanie 1,249 g pięciowodnego siarczanu miedziowego (CuSO4 x 5 H2O), czystego do analiz, z 50 ml roztworu kwasu siarkowego (H2SO4) o stężeniu 0,5 mol/l i rozcieńczeniu wodą w kolbie miarowej do objętości 1000 ml.
– Roztwór roboczy o stężeniu 0,00025 mol/l, otrzymany przez zmieszanie 50 ml roztworu podstawowego z 10 ml roztworu kwasu siarkowego (H2SO4) o stężeniu 0,5 mol/l i rozcieńczeniu wodą w kolbie miarowej do objętości 1000 ml.
m. Chlorek sodowy (NaCl), czysty do analiz.
2.2. Wymagana aparatura
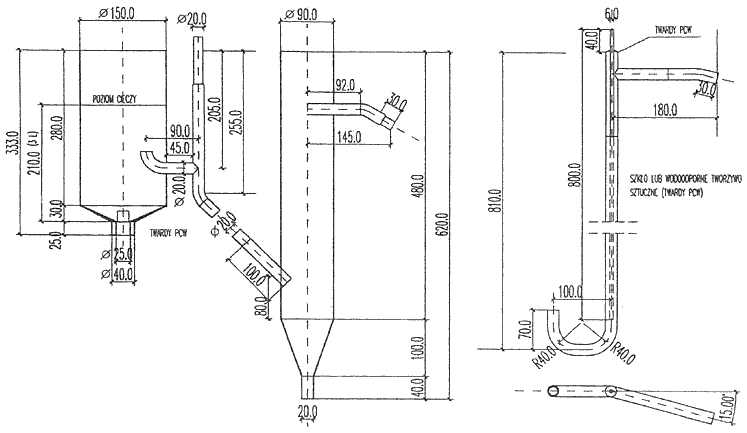
a. Aparat do zatężania i wypieniania niejonowych substancji powierzchniowoczynnych, określony w załączniku nr 8 do rozporządzenia, z zastrzeżeniem, że spiek szklany powinien mieć tę samą średnicę co część cylindryczna aparatu.
b. Mieszadło magnetyczne z mieszadełkiem o długości 25–30 mm.
c. Tygiel Goocha z dnem perforowanym o średnicy 25 mm, typ G4 lub równoważny aparat do sączenia innego typu.
d. Sączki z włókna szklanego o średnicy 27 mm i grubości włókna od 0,5 do 1,5 mm.
e. Kolba ssawkowa o pojemności 500 ml z dopasowanym kołnierzem gumowym.
f. Kolba ssawkowa o pojemności 250 ml z dopasowanym kołnierzem gumowym.
g. Potencjometr z rejestratorem wyposażonym w elektrody pomiarowe: platynową i kalomelową lub platynową i chlorosrebrową o zakresie pomiarowym 250 mV i automatyczną biuretę o pojemności 20–25 ml lub alternatywnym wyposażeniem ręcznym.
h. Rozdzielacz o pojemności 250 ml.
I Zlewki o pojemności 250 ml.
3. Wymagany sposób wykonania oznaczenia
3.1. Pobieranie próbek
Próbki nie powinny być pobierane przez warstwę piany. Próbki pobrane ze zbiornika (A) i odbieralnika (F) powinny być przefiltrowane przez bibułę filtracyjną i poddane badaniom natychmiast po pobraniu. W trakcie filtrowania należy odrzucić pierwsze 100 ml filtratu.
3.2. Zatężanie i wyplenianie niejonowej substancji powierzchniowoczynnej
Do aparatu do zatężania i wypleniania należy wprowadzić odmierzoną ilość próbki tak, aby zawierała 250–800 mg niejonowych substancji powierzchniowoczynnych. W celu ułatwienia ich wydzielenia należy dodać 100 g chlorku sodu (NaCl) i 5 g wodorowęglanu sodowego (NaHCO3).
W przypadku gdy objętość próbki przekracza 500 ml, sole te wprowadza się do aparatu w formie stałej i rozpuszcza, przedmuchując próbkę azotem.
W przypadku gdy objętość próbki jest mniejsza niż 500 ml, sole te dodaje się po uprzednim rozpuszczeniu w około 400 ml wody.
W każdym przypadku aparat należy dopełnić wodą do górnego kurka odpływowego. Na warstwę wodną wprowadzić ostrożnie 100 ml octanu etylu. Płuczkę, przez którą doprowadza się powietrze lub azot, napełnić w 2/3 pojemności octanem etylu. Przez aparaturę przepuszczać strumień gazu (azotu lub powietrza) z szybkością 30–60 l/godz., z wykorzystaniem rotametru, w celu kontroli przepływu gazu. Szybkość przepływu gazu należy zwiększać stopniowo i tak wyregulować, aby zachować wyraźny podział faz i aby na granicy faz nie powstawały zawirowania, w celu uniknięcia przechodzenia octanu etylu do fazy wodnej. Po upływie 5 minut należy zamknąć dopływ gazu. Jeżeli wskutek rozpuszczenia się w wodzie objętość fazy organicznej uległa zmniejszeniu o więcej niż o 20%, operację wypleniania należy powtórzyć, zwracając szczególną uwagę na szybkość przepływu gazu.
Fazę organiczną przenieść do rozdzielacza. Wodę, która pojawi się w rozdzielaczu z fazy wodnej, a której powinno być nie więcej niż kilka mililitrów, przenieść z powrotem do aparatu do wypieniania.
Warstwę octanu etylu należy przesączyć przez suchy sączek do zlewki o pojemności 250 ml. Do aparatu do wypleniania dodać ponownie 100 ml octanu etylu i powtórzyć trwającą 5 minut operację przepuszczania gazu. Fazę organiczną odprowadzić do tego samego rozdzielacza jak poprzednio. Fazę wodną, która ewentualnie wydzieli się, odrzucić, a fazę organiczną przesączyć przez ten sam co poprzednio, sączek i połączyć z pierwszą jej partią. Przepłukać rozdzielacz i sączek za pomocą 20 ml octanu etylu. Następnie należy odparować octan etylu do sucha na łaźni wodnej pod wyciągiem. W celu przyspieszenia parowania można skierować lekki strumień powietrza na powierzchnię roztworu.
3.3. Strącanie i filtrowanie osadu niejonowej substancji powierzchniowoczynnej
Uzyskaną, zgodnie z procedurą określoną w pkt 3.1, suchą pozostałość należy rozpuścić w 5 ml metanolu. Dodać 40 ml wody oraz 0,5 ml rozcieńczonego kwasu solnego, a następnie wymieszać całość za pomocą mieszadła magnetycznego.
Do otrzymanego roztworu dodać 30 ml odczynnika strącającego. Osad wytrąca się podczas stałego mieszania. Po 10 minutach przerwać mieszanie i pozostawić mieszaninę przez 5 minut w bezruchu. Mieszaninę przesączyć przez umieszczony na dnie aparatu filtracyjnego sączek z włókna szklanego. Sączek należy najpierw przemyć, stosując ssanie, za pomocą 2 ml lodowatego kwasu octowego.
Zlewkę, mieszadełko magnetyczne oraz tygiel należy dokładnie przemyć około 40–50 ml kwasu octowego. Ilościowe przenoszenie osadu przyczepionego do ścianek zlewki na sączek nie jest konieczne, gdyż roztwór przed miareczkowaniem przenosi się ponownie do zlewki, w której nastąpiło strącanie, i wtedy pozostały osad ulega rozpuszczeniu.
3.4. Rozpuszczanie wytrąconego osadu niejonowej substancji powierzchniowoczynnej
Osad należy rozpuścić w tyglu filtracyjnym przez dodanie gorącego, o temperaturze 80°C, roztworu winianu amonowego w trzech porcjach po 10 ml. Po dodaniu każdej porcji winianu amonowego odczekać kilka minut przed włączeniem ssania i rozpoczęciem sączenia. Zawartość kolby ssawkowej przenieść do zlewki, w której wykonano strącanie. Rozpuścić osad ze ścianek zlewki, używając 20 ml roztworu winianu amonowego. Dokładnie przemyć tygiel, kołnierz gumowy i kolbę ssawkową, stosując 100–150 ml wody i zawracając ją do zlewki stosowanej do strącania.
3.5. Miareczkowanie
Mieszając roztwór za pomocą mieszadła magnetycznego, należy dodać kilka kropli roztworu purpury bromokrezolowej, a następnie roztwór amoniaku, aż do uzyskania zmiany barwy roztworu na fioletową (wyjściowy roztwór jest słabo kwaśny z powodu obecności resztek kwasu octowego użytego do przemycia).
Następnie należy dodać 10 ml standardowego roztworu buforowego, zanurzyć elektrody w roztworze 1 przy zanurzonym końcu biurety miareczkować układ potencjometrycznie za pomocą roztworu pirolidynoditiokarbaminianu sodowego, zapisując kolejne wskazania potencjometru. Miareczkowanie należy prowadzić aż do momentu przekroczenia skoku potencjału. Szybkość miareczkowania nie powinna przekraczać 2 ml/min. Za punkt końcowy miareczkowania przyjmuje się punkt przecięcia stycznych do obu ramion krzywej zmiany potencjału.
Uwaga:
Obserwowane niekiedy spłaszczenie skoku potencjału można wyeliminować przez lekkie wygładzenie elektrody platynowej papierem ściernym.
3.6. Ślepa próba
Równolegle z właściwym oznaczeniem powinno wykonać się ślepą próbę, stosując układ składający się z 5 ml metanolu i 40 ml wody. Analizę należy przeprowadzić analogicznie jak analizę próbki badanej.
3.7. Sprawdzanie miana roztworu pirolidynoditiokarbaminianu sodowego
Miano roztworu pirolidynoditiokarbaminianu sodowego powinno być sprawdzone w dniu użycia.
W tym celu należy zmieszać 10 ml wzorcowego roztworu roboczego siarczanu miedziowego z 100 ml wody i 10 ml standardowego buforu octanowego, a następnie zmiareczkować całość.
Miano roztworu pirolidynoditiokarbaminianu sodowego f oblicza się z wzoru:
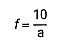
w którym:
a – oznacza objętość roztworu pirolidynoditiokarbaminianu sodowego zużytego do miareczkowania, w ml.
4. Obliczanie wyniku
Stężenie niejonowych substancji powierzchniowo czynnych w przeliczeniu na nonylofenol oksyetylenowany 10 molami tlenku etylenu XBiAS, w mg/l, oblicza się wg wzoru:
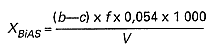
w którym:
b – oznacza objętość roztworu pirolidynoditiokarbaminianu sodowego zużytego do miareczkowania próbki w ml,
c – oznacza objętość roztworu pirolidynoditiokarbaminianu sodowego zużytego do miareczkowania ślepej próbki w ml,
f – oznacza miano roztworu pirolidynoditiokarbaminianu sodowego,
V – oznacza objętość próbki pobranej do badań w ml,
0,054 – oznacza współczynnik przeliczeniowy.
Wynik oznaczenia podaje się, jako średnią arytmetyczną wyników co najmniej dwóch wyników oznaczeń, z dokładnością do 0,1 mg/l BiAS.
W zależności od miejsca pobrania próbki XBiAS stanowi odpowiednio D lub O przy obliczaniu dziennej wartości stopnia biodegradacji,
gdzie:
D – oznacza stężenie anionowych lub niejonowych substancji powierzchniowoczynnych w tzw. dopływie (oznaczanych odpowiednio jako MBAS lub BiAS) w mg/l,
O – oznacza stężenie anionowych lub niejonowych substancji powierzchniowoczynnych w tzw. odpływie (oznaczanych odpowiednio jako MBAS lub BiAS) w mg/l.
Załącznik nr 8
SCHEMAT APARATURY WYMAGANEJ DO ZATĘŻANIA l WYPIENIANIA NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
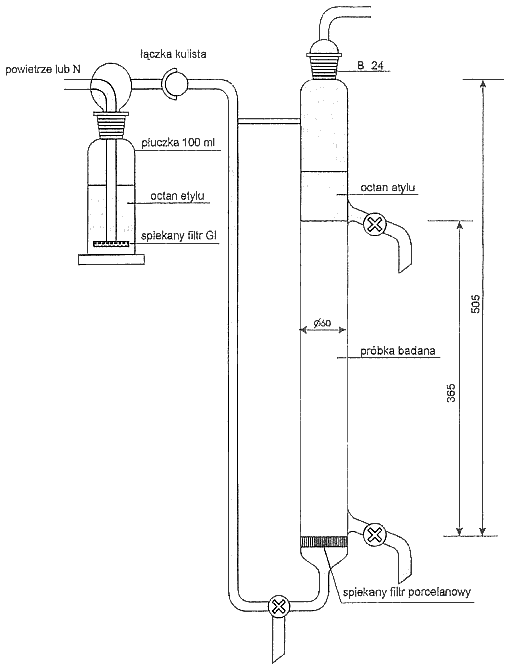
Załącznik nr 9
METODYKA OBLICZANIA STOPNIA BIODEGRADACJI ANIONOWYCH l NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH
1. Zasada metody
Metoda polega na obliczaniu dziennych wartości stopnia biodegradacji na podstawie stężeń odpowiednio anionowych i niejonowych substancji powierzchniowoczynnych w dopływie, tj. w cieczy wprowadzanej do komory napowietrzania osadu czynnego, będącej ściekiem syntetycznym zawierającym badane substancje powierzchniowoczynne oraz w odpływie, tj. w cieczy odprowadzanej z komory napowietrzania do odbieralnika po procesie osadzania osadu czynnego, a następnie na obliczaniu stopnia biodegradacji anionowych i niejonowych substancji powierzchniowoczynnych, będącego średnią arytmetyczną dziennych wartości stopnia biodegradacji po zakończeniu okresu wstępnej adaptacji osadu czynnego.
2. Wymagany sposób obliczania dziennej wartości stopnia biodegradacji
Dzienną wartość stopnia biodegradacji anionowych lub niejonowych substancji powierzchniowoczynnych oblicza się jako iloraz różnicy stężeń anionowych lub niejonowych substancji powierzchniowoczynnych w tzw. dopływie i w tzw. odpływie oraz zawartości anionowych lub niejonowych substancji powierzchniowoczynnych w tzw. dopływie.
Dzienną wartość stopnia biodegradacji Bi, w %, określa się według wzoru:
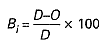
w którym:
D – oznacza stężenie anionowych lub niejonowych substancji powierzchniowoczynnych w tzw. dopływie (oznaczanych odpowiednio jako MBAS lub BiAS) w mg/l,
O – oznacza stężenie anionowych lub niejonowych substancji powierzchniowoczynnych w tzw. odpływie (oznaczanych odpowiednio jako MBAS lub BiAS) w mg/l.
Dzienną wartość stopnia biodegradacji podać z dokładnością do 0,1%.
3. Wymagany sposób obliczania stopnia biodegradacji anionowych i niejonowych substancji powierzchniowoczynnych
Stopień biodegradacji anionowych i niejonowych substancji powierzchniowoczynnych stanowi średnią arytmetyczną dziennych wartości stopnia biodegradacji otrzymanych w ciągu 21 dni równomiernej, przebiegającej bez zakłóceń pracy urządzenia, po okresie adaptacji osadu czynnego. Czas adaptacji w żadnym wypadku nie może przekroczyć 6 tygodni.
Dopuszcza się zmniejszenie liczby próbek pobieranych w ciągu 21 dni następujących po zakończeniu okresu wstępnej adaptacji osadu czynnego, jednak dla obliczenia wartości średniej powinno ich być co najmniej 14.
Stopień biodegradacji B (%) anionowych i niejonowych substancji powierzchniowoczynnych oblicza się wg wzoru:
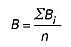
w którym:
n – oznacza liczbę oznaczeń.
Stopień biodegradacji anionowych i niejonowych związków powierzchniowoczynnych podać z dokładnością do 1%.
Wyniki należy podać w formie sprawozdania, w którym zamieszcza się dane laboratorium wykonującego badania, dane o próbce, datę rozpoczęcia i zakończenia badań, dzienne wartości stopnia biodegradacji oraz stopień biodegradacji odpowiednio anionowych i niejonowych substancji powierzchniowoczynnych oraz uwagi o przebiegu analizy. Wyniki należy przedstawić również w formie graficznej.
Załącznik nr 10
WYKRES ILUSTRUJĄCY WYMAGANY PRAWIDŁOWY PRZEBIEG PROCESU BIODEGRADACJI ANIONOWYCH l NIEJONOWYCH SUBSTANCJI POWIERZCHNIOWOCZYNNYCH W CZASIE
- Data ogłoszenia: 2002-11-26
- Data wejścia w życie: 2002-12-11
- Data obowiązywania: 2002-12-11
- Dokument traci ważność: 2007-10-10

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA